我一直强调:数据挖掘的核心是缩小目标基因!
各种数据挖掘文章本质上都是要把目标基因集缩小,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学算法,最后搞成10个左右的基因组成signature即可顺利发表。
其实还有另外一个策略方向,有点类似于人工选择啦,通常是可以往热点靠,比如肿瘤免疫,相当于你不需要全部的两万多个基因的表达量矩阵进行后续分析,仅仅是拿着几千个免疫相关基因的表达矩阵即可。最近比较热门的有:自噬基因,铁死亡,EMT基因,核受体基因家族,代谢基因。还有一个最搞笑的是m6a基因的策略,完全是无厘头的基因集搞小,纯粹是为了搞小而搞小。
虽然说数据挖掘的核心就是把基因集的数量搞小,但是实际上大家都只是在自己的数据集上面自圆其说而已。最近听一个演讲,就提到了在乳腺癌领域的已经发表的33个基因集,他们的overlap其实非常少,如下所示:
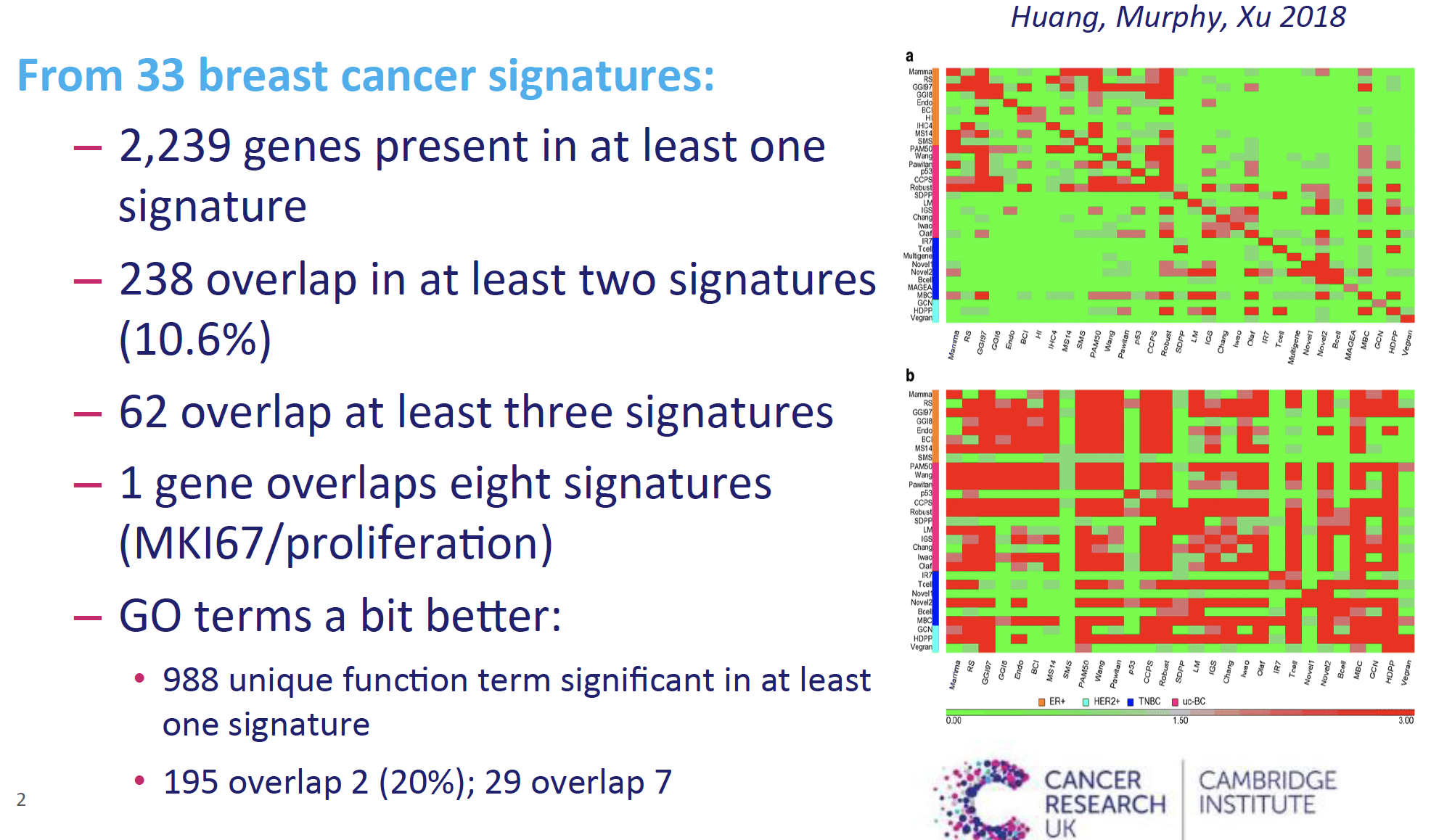
绝大部分的基因仅仅是单一的基因集里面出现,不具备可重复性。出现两次及以上的也仅仅是10%的基因,出现在8个以上的基因集里面的基因就只剩下1个了,就是MKI67代表细胞增殖功能的,更别说是出现在33个基因集里面的。
但是,如果考虑这些基因所 代表的GO数据库功能条目,重复性就好了一点,至少出现2次的基因提高到了20%。
然后研究者单独拿出来了乳腺癌领域久经考验的70个基因 的基因集,发现如果是随机选择70个基因,随机1万次,也有接近30%的可能性拿到比这个久经考验的70个基因 的基因集表现要好的情况。

为什么会出现这样的情况呢?
其实如果你拿到任何一个带有临床生存信息表达矩阵,都可以拿到很多有统计学意义的生存相关基因,这些基因都可以找到共表达基因,它们本来就相互连接起来了。
学徒作业
就拿TCGA数据库的乳腺癌的RNA-seq矩阵,从ucsc的xena浏览器下载,1000个左右的肿瘤病人数据里面的全部的基因做wgcna后看看分成多少个模块。
然后看看统计学显著的2000多个生存相关基因再次wgcna分成多少个模块。
然后看看两个模块根据各自的数据库功能能不能对应起来。
历年学徒作业目录如下:
- 生信编程直播课程优秀学员作业展示1
- 生信编程直播课程优秀学员学习心得及作业展示3
- 生信编程直播课程优秀学员作业展示2
- 给学徒的GEO作业
- 这个WGCNA作业终于有学徒完成了!
- 上次说的gmt函数(学徒作业)
- 拖后腿学徒居然也完成作业,理解RNA-seq数据分析结果
- 肿瘤外显子视频课程小作业
- ChIPseq视频课程小作业
- Agilent芯片表达矩阵处理(学徒作业)
- 学徒作业:TCGA数据库单基因gsea之COAD-READ
- 学徒作业-在CCLE数据库里面根据指定基因在指定细胞系里面提取表达矩阵
- 学徒作业-指定基因在指定组织里面的表达量热图
- 学徒作业-我想看为什么这几个基因的表达量相关性非常高
- 学徒作业:给你8个甲基化探针, 你在tcga数据库进行任意探索
- 学徒作业-根据我的甲基化视频教程来完成2015-NPC-methy-GSE52068研究
- RNA芯片和测序技术的比较(学徒作业)
- 学徒作业-单基因的tcga数据挖掘分析
- ATCC终于出来了organoids资源
- 拿到7个DDR通路的基因集-学徒作业
- 绘图本身很简单但是获取数据很难
- 都说lncRNA只有部分具有polyA尾结构,请证明
- 学徒作业-hisat2+stringtie+ballgown流程
- 学徒任务-探索DNA甲基化的组织特异性
- 用WES和RNA-Seq数据提取到的somatic SNVs不一致
- 《GEO数据挖掘课程》配套练习题