前面我们的学徒作业:学徒任务-探索DNA甲基化的组织特异性,大家完成的不多,可能是甲基化芯片数据处理对大家来说不紧急也不必须吧。不过最近刷文献看到了,另外一个策略,可以做多分组的甲基化差异分析,而不是一对多的差异分析策略。
是一个Java软件可以做quantitative differentially methylated regions (QDMRs) ,发表在 2011 Feb 8. doi: 10.1093/nar/gkr053,是Nucleic Acids Res. 杂志。甲基化技术
主要是 ,甲基化测序的 WGBS和RRBS,还有 芯片:
- 全基因组DNA甲基化测序(Whole Genome Bisulfite Sequencing,WGBS)是 DNA 甲基化研究的金标准,它通过 Bisulfite 处理和全基因组 DNA 测序结合的方式,对整个基因组上的甲基化情况进行分析,具有单碱基分辨率,可精确评估单个 C 碱基的甲基化水平,构建全基因组精细甲基化图谱。数据量非常大。
- 简化甲基化测序 (Reduced representation bisulfite sequencing, RRBS)是一种准确、高效、经济的DNA甲基化研究方法,通过酶切 (Msp I) 富集启动子及CpG岛区域,并进行Bisulfite测序,同时实现DNA甲基化状态检测的高分辨率和测序数据的高利用率。作为一种高性价比的甲基化研究方法,简化甲基化测序在大规模临床样本的研究中具有广泛的应用前景。
- Illumina的Infinium BeadChip芯片,包括HumanMethyation450(450K)和MethylationEPIC(850K)。Infinium芯片存在染料偏差、不同探针化学和位置效应的问题,已知这些问题会影响结果,必须在数据处理过程中进行校正。Infinium 450K探针交叉反应和模糊比对到人类基因组中的多个位置影响了485,000个探测器中的约140,000个探针(29%),将可用探针的数量减少到约345,000个。这个问题在新发布850K仍然存在,其包括> 90%的450K探针。
有文章比较这3个技术:Empirical comparison of reduced representation bisulfite sequencing and Infinium BeadChip reproducibility and coverage of DNA methylation in humans,感兴趣的都是可以自由阅读,提高自己哈。
甲基化信号的主要分析也是 差异甲基化区域(DMRs)与 DMR 相关差异基因。功能区域分类
不同区域的甲基化位点信号值的生物学意义完全不一样,很多研究都喜欢混淆它们,我觉得有必要重点指出来:
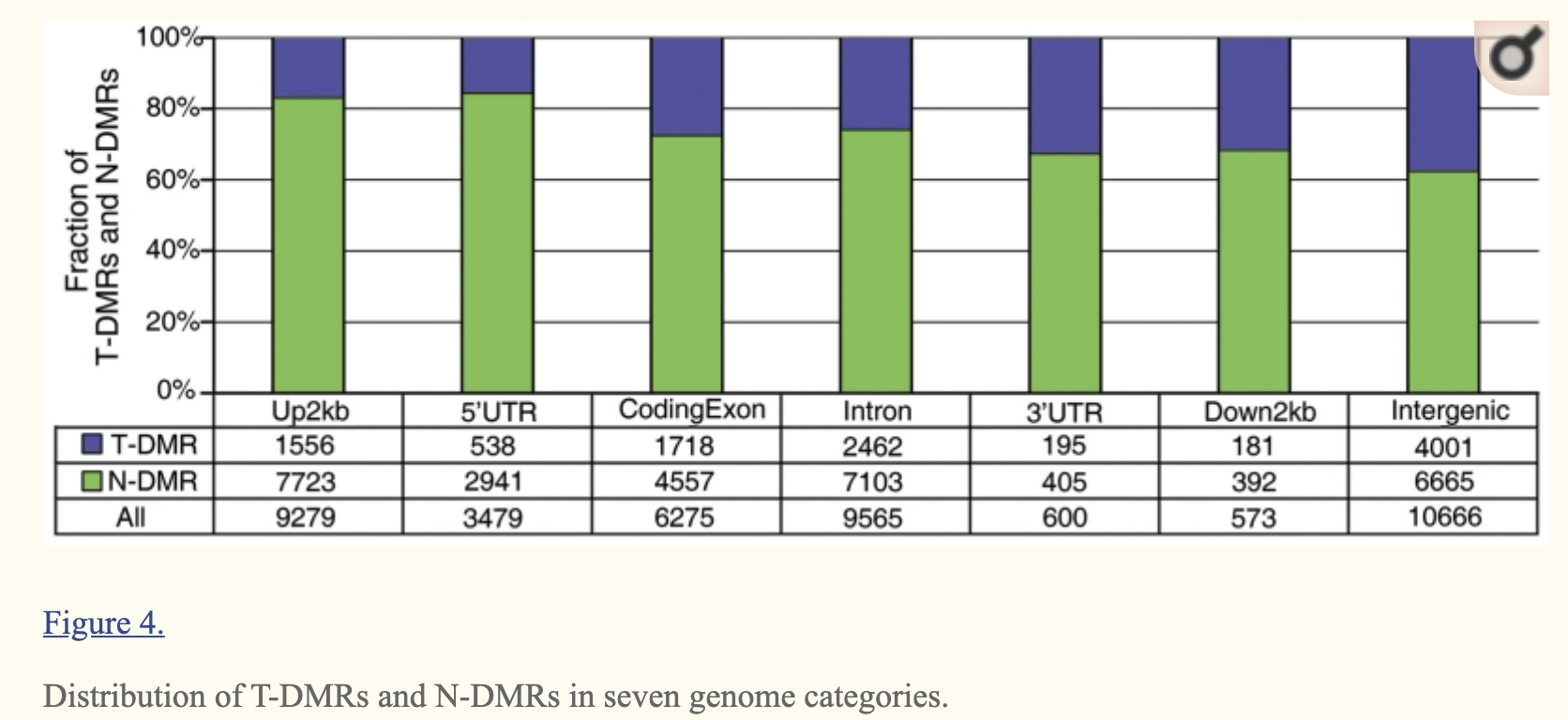
分类:(Up2kb, 5′-UTR, CodingExon, Intron, 3′-UTR, Down2kb and Intergenic regions)
比如就是doi: 10.1093/nar/gkr053文章就是:
实例
看到发表在2018的文章《Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns》就是通过对TCGA数据库的BRCA的甲基化芯片数据和RNA-seq数据联合分析:
- 甲基化芯片是450K,数据预处理阶段仅保留了TSS附近的位点。
- 转录组测序数据是RSEM的表达矩阵,也使用了combat去除批次效应
- 根据KM和COX两个生存分析来挑选到了3869个生存相关的甲基化探针
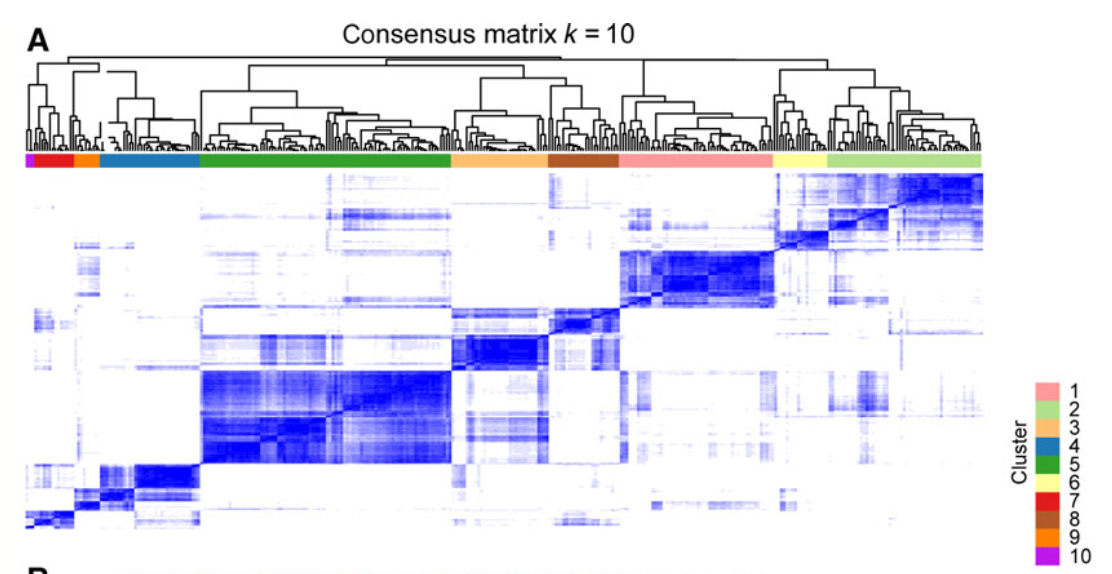
- 对3869个甲基化探针在669个病人的信号值矩阵使用R包ConcensusClusterPlus进行聚类分群,定为9群。
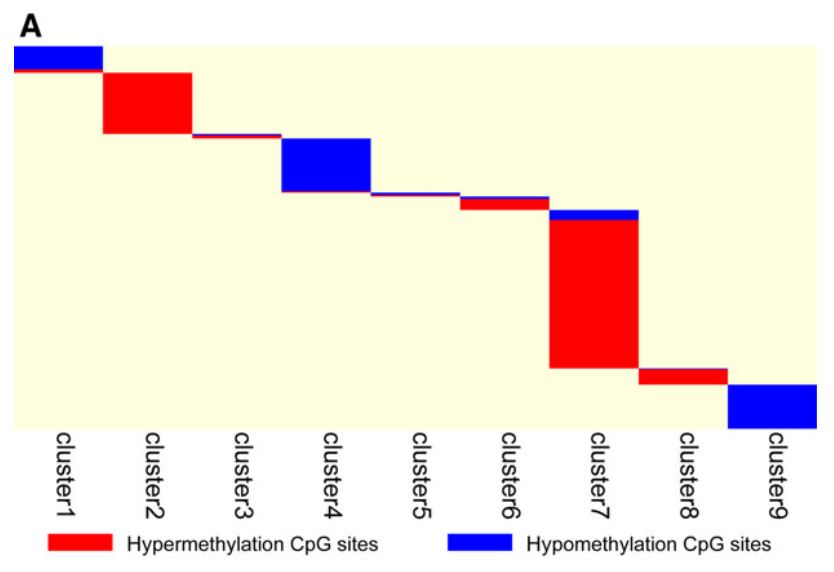
首先当然是比较了生存相关甲基化探针的10群和乳腺癌领域众所周知的PAM50分类进行对比,然后找寻每个亚群的特异性甲基化探针。
找寻每个亚群的特异性甲基化探针的方法如下:
可以看到,就是使用了我们最开始提到的Java软件可以做quantitative differentially methylated regions (QDMRs) ,发表在 2011 Feb 8. doi: 10.1093/nar/gkr053,是Nucleic Acids Res. 杂志。
结果图清晰明了:
需要注意的是,之前的生存相关的3869个甲基化探针,被这个分析过滤了一下后,成为了1252个探针哦。
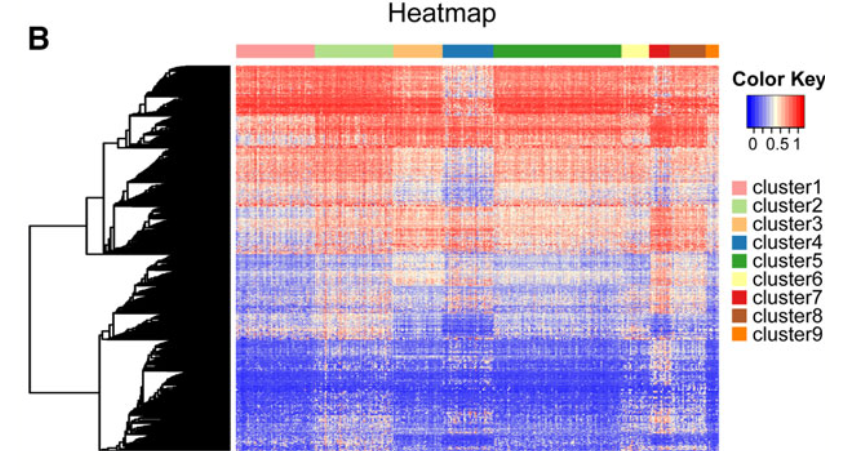
而且,单独提前这1252个甲基化探针的信号值,去669个病人里面,看信号值热图,可以看到,很清晰的9个亚群:
这个是必然滴,因为前面就是通过它的生存相关的3869个甲基化探针使用R包ConcensusClusterPlus进行聚类分群的, 这次单独筛选了更特异性的,只能说让分群更完美!
生信门槛
这样的研究门槛主要是在数据整理阶段,这个示例里面就是TCGA数据库的整理啦,筛选到合适的甲基化芯片数据已经转录组测序数据,然后生存分析等等,一些R包的学习。
再怎么强调生物信息学数据分析学习过程的计算机基础知识的打磨都不为过,我把它粗略的分成基于R语言的统计可视化,以及基于Linux的NGS数据处理:- 《生信分析人员如何系统入门R(2019更新版)》
- 《生信分析人员如何系统入门Linux(2019更新版)》
把R的知识点路线图搞定,如下:- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班:- 数据挖掘学习班第7期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第9期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外,:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》