我们的CNS图表复现之旅已经开始,上一讲是:
如果你也想加入交流群,自己去:你要的rmarkdown文献图表复现全套代码来了(单细胞)找到我们的拉群小助手哈。
文章的第一次分群拿到了epithelial cells [n = 5,581], 我们的CNS图表复现拿到的上皮细胞数量是:
> table(sce@meta.data$immune_annotation)
epi immune stromal
5444 13792 4278
我在单细胞天地的教程:是否是免疫细胞很容易区分那是否是肿瘤细胞呢?提到过,常规做法是使用inferCNV算法可以区分细胞恶性与否。比如online 29 April 2020的文章《Single-Cell Transcriptome Analysis Reveals Intratumoral Heterogeneity in ccRCC, which Results in Different Clinical Outcomes》,就是选取ccRCC的3个病例的21个样本(12个肿瘤,9个对照),质控后总计24550个细胞,使用inferCNV算法可以区分成为7786个非恶性,16764个恶性细胞。但其实我是提出来了一个开放式问题共大家讨论,除了这个inferCNV算法,难道就没有其它方法来比较好的区分细胞的恶性与否吗?希望大家集思广益吧!
现在就给大家演示一下,不使用inferCNV算法仍然是可以判断上皮细胞的恶性与否的方法。
首先取上皮细胞的子集
关键就是 subset 函数的使用:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
load(file = 'first_sce.Rdata')
load(file = 'phe-of-first-anno.Rdata')
sce=sce.first
table(phe$immune_annotation)
sce@meta.data=phe
cells.use <- row.names(sce@meta.data)[which(phe$immune_annotation=='epi')]
length(cells.use)
sce <-subset(sce, cells=cells.use)
sce
然后重新走单细胞流程:
完全参考单细胞转录组数据分析的流程:
# Epithelial cells (n = 5,581) were subsetted and re-clustered into 26 discrete epithelial clusters
sce <- NormalizeData(sce, normalization.method = "LogNormalize",
scale.factor = 1e6)
GetAssay(sce,assay = "RNA")
sce <- FindVariableFeatures(sce,
selection.method = "vst", nfeatures = 2000)
# 步骤 ScaleData 的耗时取决于电脑系统配置(保守估计大于一分钟)
sce <- ScaleData(sce)
sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
DimHeatmap(sce, dims = 1:12, cells = 100, balanced = TRUE)
ElbowPlot(sce)
sce <- FindNeighbors(sce, dims = 1:15)
# Epithelial cells (n = 5,581) were subsetted and re-clustered into 26 discrete epithelial clusters
res.used <- 1
sce <- FindClusters(object = sce, verbose = T, resolution = res.used)
table(sce@meta.data$seurat_clusters)
检查单细胞分群及其来源于的病人信息
很简单的可视化:
set.seed(123)
sce <- RunTSNE(object = sce, dims = 1:15, do.fast = TRUE)
DimPlot(sce,reduction = "tsne",label=T)
DimPlot(sce,reduction = "tsne",label=T, group.by = "patient_id")
table(sce@meta.data$seurat_clusters)
library(gplots)
tab.1=table(sce@meta.data$seurat_clusters,sce@meta.data$patient_id)
balloonplot(tab.1)
可以很明显的看到,第1,2,7,14,21,23,25 是跨越病人的聚类情况,如下所示:
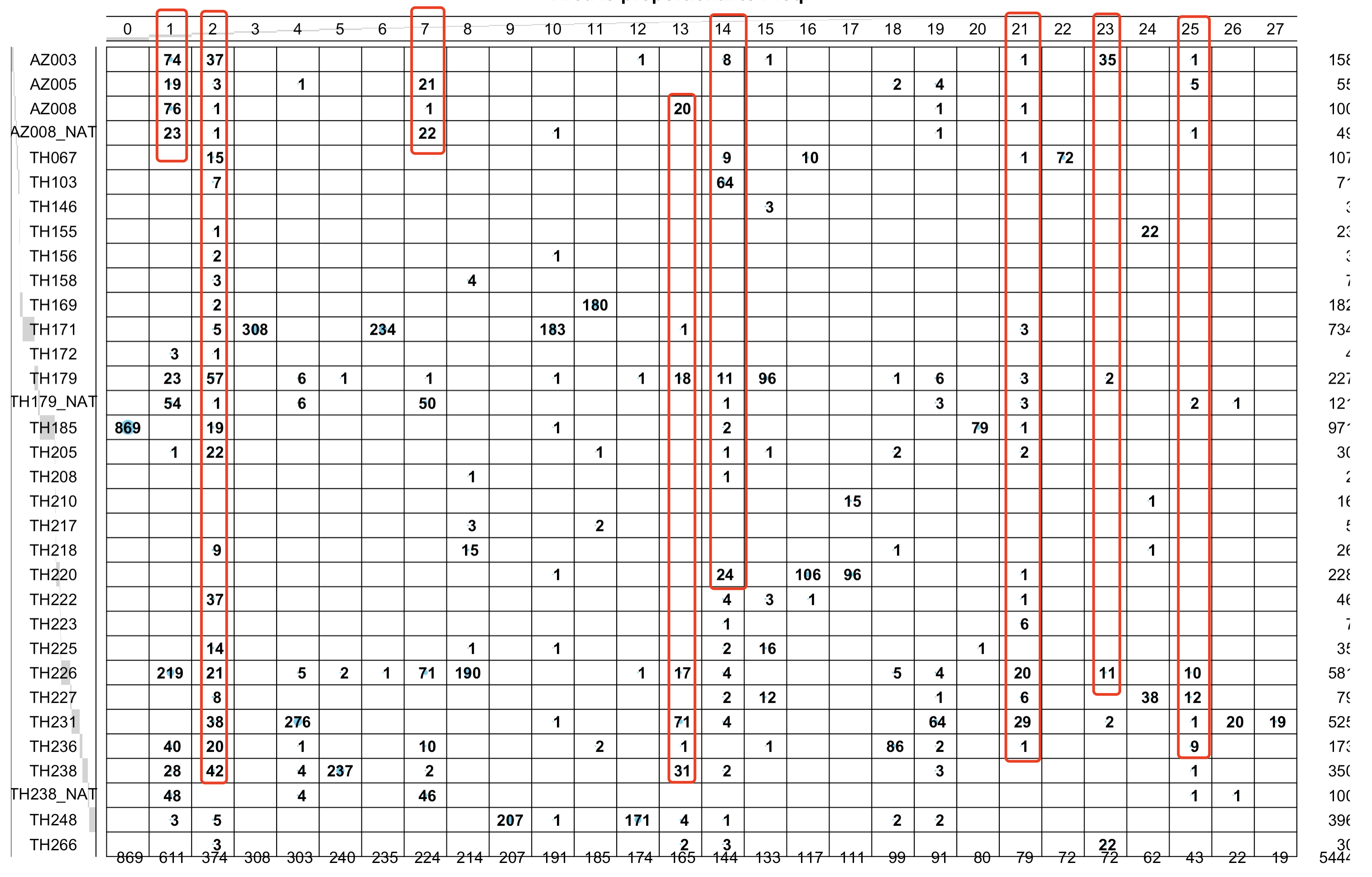
根据细胞亚群是否跨越病人存在来区分上皮细胞的恶性与否
代码如下:
# 1,2,7,14,21,23,25 是跨越病人的聚类情况
# Cancer cells were identified in 44 of the original 49 tumor biopsy samples
# All cancer cells (n = 3,754) were re-clustered, resulting in 25 unique clusters (Figures S2A and S2B).
# The non-cancer epithelial cell clusters (n = 16) were further annotated into cell subtype
cancer=c(1,2,7,13,14,21,23,25)
sce@meta.data$cancer <-ifelse(sce@meta.data$seurat_clusters %in% cancer ,'non-cancer','cancer')
# MAke a table
table(sce@meta.data$cancer)
phe=sce@meta.data
可以看到,我们得到的恶性细胞数量跟作者文章的一样的:
> table(sce@meta.data$cancer)
cancer non-cancer
3732 1712