前面我分享了:最新最全的mutect2教程,提到了其实大家不必在一棵树上吊死,GATK的Mutect2流程跑不通就换一个软件咯,2018年文章:A review of somatic single nucleotide variant calling algorithms fornext-generation sequencing data 就囊括了十几款找somatic mutation的软件。当然了,绝大部分软件其实是没有尝试的价值。不过如果要是从安装和使用的简易性来考虑,varscan 软件必须值得一提。
我在生信技能树发布的很多关于varscan 软件找somatic mutation教程都过时了,如下:
毕竟是三年多过去了。不过,varscan相比起GATK的Mutect2流程的更新频率来说,就是小巫见大巫了。正好我放弃了GATK的Mutect2流程,就使用varscan 软件找somatic mutation,我可以保证,这个教程应该是够用十年了,因为varscan 软件已经停止更新了。
背景知识
体细胞突变(somatic mutation)是指患者某些组织或者器官后天性地发生了体细胞变异,虽然它不会遗传给后代个体,却可以通过细胞分裂,遗传给子代细胞。体细胞突变对肿瘤的发生发展有关键性的作用,并且它也是制定肿瘤癌症靶向治疗措施的关键所在。
NGS使体细胞变异的检测更加全面,成本更低,在检测多种体细胞变异上具有很大的优势,但在使用过程中还存在着挑战:如样品降解、覆盖度不足、遗传异质性和组织污染(杂质)等问题。 为应对以上挑战,降低错误率,科学家采取了不同的算法和统计模型用于检测体细胞突变。目前最受欢迎的有Varscan、SomaticSniper、 Strelka 和MuTect2 。这些软件大都是直接对肿瘤-正常样本的每个位点进行比较,对肿瘤样本中明显高于正常样本的次等位基因进行标记,作为体细胞变异,同时排除种系突变和杂合性丢失(LOH)情况。虽然这些软件具有较高的引用率,并在不断地更新,但仍存在不足:
-
a 、缺乏完整可靠的实验来评估检测结果;
-
b、 缺乏金标准,不能保证检测到的灵敏度和特异性最高;
-
c、 在实际应用中,各软件的相对优缺点在很大程度上是未知的。
下面是TCGA计划采取的体细胞突变(somatic mutation)检测软件:
- MuSE
- varscan
- MuTect
- SomaticSniper
大家可以去下载到TCGA计划的这4个软件输出的maf文件格式的somatic突变信息文件哦。
首先下载安装 varscan软件
软件发表在 2013 Dec 12. doi: 10.1002/0471250953.bi1504s44,文章标题是;Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection
软件本身功能有3个,如下 :
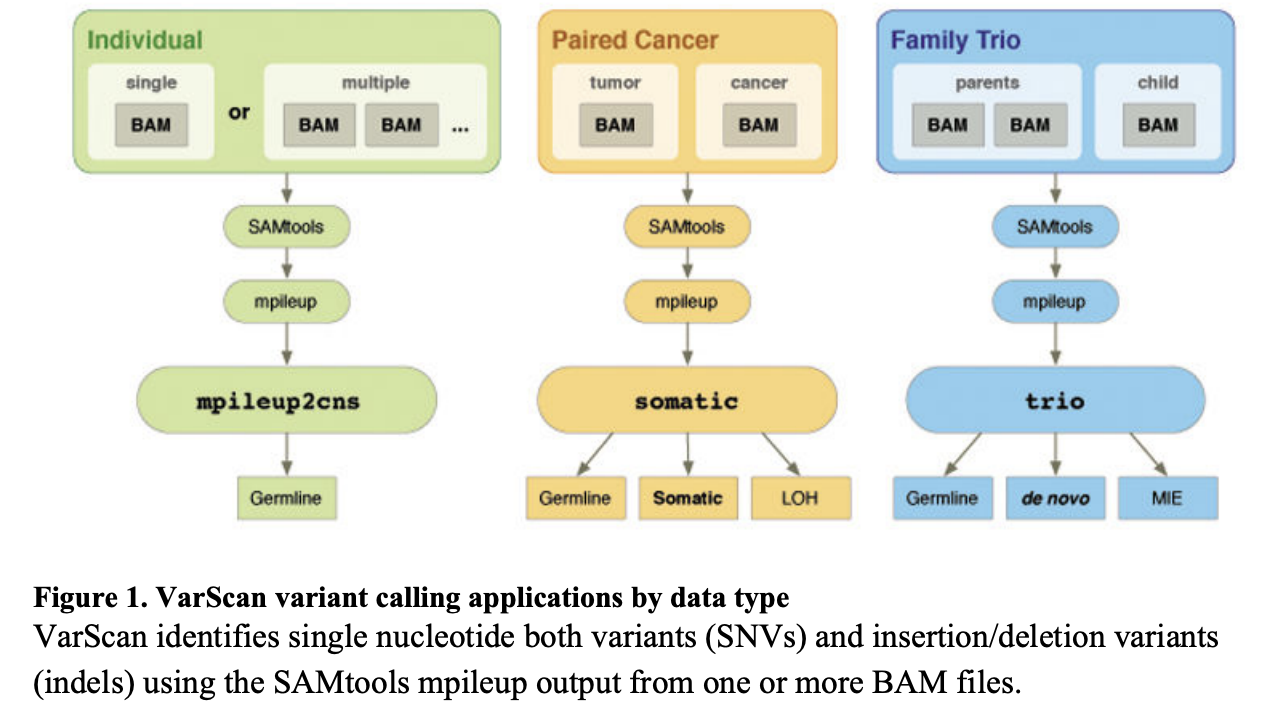
不过,我们主要是看它的找somatic mutation模式,需要肿瘤病人配对的两个测序数据的bam文件哦。
肿瘤配对样品运行VarScan的
记住:需要肿瘤病人配对的两个测序数据的bam文件,这个是找somatic突变位点的前提条件。
我写了一个脚本方便批量运行,run_varscan.sh 脚本需要4个参数,运行起来很简单。
其中最重要的参数就是一个config文件,主要是3列信息:
- 第一列是肿瘤命名
- 第二列是肿瘤病人的normal组织的bam文件地址
- 第三列是肿瘤病人的肿瘤组织的bam文件地址。
这个config文件在我的run_varscan.sh 脚本的第2位参数上面,全部代码如下:
analysis_dir=$1
config_file=$2
number1=$3
number2=$4
# for i in {0..5};do ( nohup bash run_varscan.sh ./ config 6 $i 1>$i.log 2>&1 & );done
# ps -ef |grep run_varscan.sh |awk '{print $2}' |xargs kill
reference=$HOME/biosoft/GATK/resources/bundle/mm10/Sequence/WholeGenomeFasta/genome.fa
# 不同物种的参考基因组不一样,在得到bam文件的时候就需要注意,一定要统一
cat $config_file |while read id
do
arr=($id)
normal_bam=${arr[1]}
tumor_bam=${arr[2]}
sample=${arr[0]}
if((i%$number1==$number2))
then
# A1271-1_varscan.snp.Somatic.hc
if [ ! -f ${sample}_varscan.snp.Somatic.hc.vcf ]; then
start=$(date +%s.%N)
echo VarScan `date`
normal_pileup="samtools mpileup -q 1 -f $reference $normal_bam";
tumor_pileup="samtools mpileup -q 1 -f $reference $tumor_bam";
# Next, issue a system call that pipes input from these commands into VarScan :
java -jar ~/biosoft/VarScan/VarScan.v2.3.9.jar \
somatic <($normal_pileup) <($tumor_pileup) ${sample}_varscan --output-vcf
java -jar ~/biosoft/VarScan/VarScan.v2.3.9.jar processSomatic ${sample}_varscan.snp.vcf
java -jar ~/biosoft/VarScan/VarScan.v2.3.9.jar processSomatic ${sample}_varscan.indel.vcf
echo VarScan `date`
dur=$(echo "$(date +%s.%N) - $start" | bc)
printf "Execution time for VarScan : %.6f seconds" $dur
echo
fi
fi
i=$((i+1))
done
每个样品都会输出很多文件,但是其实我们最关心的仅仅是snp.Somatic.hc这个文件而已。 上面的代码看起来很复杂,但是抛开shell脚本后,软件用法其实就两句话,somatic命令+processSomatic命令而已。
到此为止,varscan 软件找somatic mutation的流程就完成啦。但是找到了somatic mutation仅仅是万里长征的第一步,后续如何去对找到的somatic mutation进行各种各样的注释才是重点。
vcf文件转为maf格式
对somatic mutation来说,vcf格式仅仅是开始,必须转为maf格式才能做后续分析。
这里推荐使用conda来安装vcf2maf和vep两个软件,代码如下:
conda create -n vep -y
conda activate vep
conda install -y -c bioconda vcf2maf
conda install -y -c bioconda ensembl-vep
conda remove samtools
conda install -y -c bioconda samtools
vcf2maf.pl --hlep
vep --help
samtools -v
perl -e '{print join"\n",@INC}'
export VEP_PATH=$HOME/vep
export VEP_DATA=$HOME/.vep
mkdir -p $VEP_PATH $VEP_DATA; cd $VEP_PATH
vep_install -a cf -s homo_sapiens -y GRCh38 -c $VEP_PATH --CONVERT
ref=$HOME/biosoft/GATK/resources/bundle/hg38/Homo_sapiens_assembly38.fasta
vcf2maf.pl --input-vcf T1520021_varscan.snp.Somatic.hc.vcf \
--output-maf test.maf --normal-id NORMAL --tumor-id TUMOR \
--ref-fasta $ref \
--vep-data $HOME/vep \
--vep-path ~/miniconda3/envs/vep/bin/ \
--ncbi-build GRCh38
如果是多个vcf文件,就需要写脚本批量运行啦。
附上TCGA数据库maf突变资料官方大全
因为TCGA计划跨时太长,这些年找somatic变异的软件也很多,所以TCGA团队下功夫在计划结束后(April 2018)完整的系统性的整理了最后的somatic突变数据。依托于文章:Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines March 201810.1016/j.cels.2018.03.002
纳入的软件包括:
| Deposited Data | ||
|---|---|---|
| MC3 Files | https://gdc.cancer.gov/about-data/publications/mc3-2017 | |
| Software and Algorithms | ||
| MuTect | https://github.com/broadinstitute/mutect | |
| Pindel | https://github.com/genome/pindel | |
| Radia | https://github.com/aradenbaugh/radia | |
| VarScan2 | http://dkoboldt.github.io/varscan/ | |
| SomaticSniper | https://github.com/genome/somatic-sniper | |
| MuSE | https://github.com/danielfan/MuSE | |
| Indelocator | http://archive.broadinstitute.org/cancer/cga/indelocator | |
| Maf2Vcf | https://github.com/covingto/vcf2maf/ |
全部样本的somatic变异文件合并起来是七百多M,MC3 Public MAF - mc3.v0.2.8.PUBLIC.maf.gz