在2010-2015年间,RNA-seq本身就是跟现在的单细胞差不多的当红炸子鸡的地位,无数的软件工具,网页数据库,测评文章涌现出来。很多课题组导师都认为做一个RNA-seq项目就能发CNS啦,就跟这两年大家以为做一个单细胞转录组项目就可以发CNS的坚信程度是一模一样的!
直到现在(2020),基于高通量测序技术的RNA-Seq方法仍然是转录组学研究中必不可少的工具。截止到(2016)已经普遍接受的是,标准化预处理步骤可以显着提高分析质量,特别是对于差异基因表达分析而言。 然而,彼时尚未找到金标准归一化方法。我在生信技能树的教程呢,通常是直接就推荐3大R包(limma,edgeR,DEseq2),转录组的基本分析教程合辑:
-
上游分析视频以及代码资料在:https://share.weiyun.com/5QwKGxi
-
下游主要是基于counts矩阵的标准分析的代码 https://share.weiyun.com/50hfuLi
很多人就问我这样推荐的理由,有没有参考文献,但是前些日子一直比较忙,就没有回复大家。恰好最近整理我五年前收集的RNA-seq资料,重新发现了一个能比较好支持3大R包(limma,edgeR,DEseq2)的文献。
文章详情:Maza E (2016) In Papyro Comparison of TMM (edgeR), RLE (DESeq2), and MRN Normalization Methods for a Simple Two-Conditions-Without-Replicates RNA-Seq Experimental Design. Front Genet 7:164. [article]
一图概况如下:
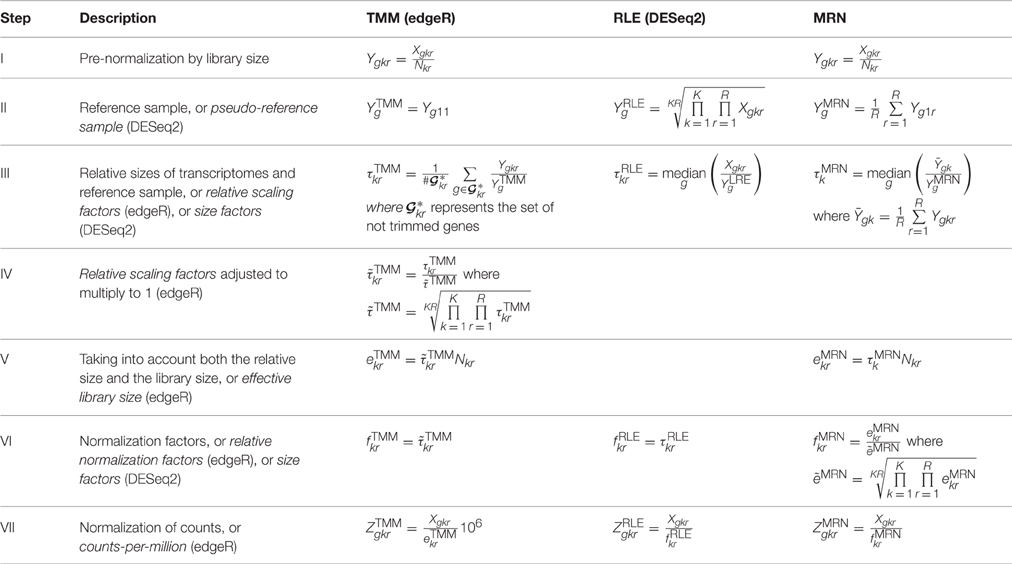
文章提到了以下3个算法,做了一下测试数据的比较:
- The first method is the “Trimmed Mean of M-values” normalization (TMM) described in and implemented in the edgeR package.
- The second method is the “Relative Log Expression” normalization (RLE) implemented in the DESeq2 package.
- The third method is the “Median Ratio Normalization” (MRN).
作者的测试数据是:a matrix of counts: 34675 rows (genes) and 9 columns (samples from 3 stages and 3 biological replicates per stage). 一个 in silico calculations carried out on a given real data set from the tomato fruit set.
作者的结论很有意思:
- For a very simple experimental design, i.e., about two conditions and no replicates, users can use any of the three studied normalization methods with no impact on results.
- But, for a more complex experimental design, the MRN method could be adopted.
学徒作业,以仅提供bam文件的RNA-seq项目重新分析 教程提到的数据集为例子,比较3大R包(limma,edgeR,DEseq2)差异分析的结果,绘制一个韦恩图或者其它可视化的展现形式!因为这个RNA-seq项目的数据库链接在:https://www.ebi.ac.uk/ena/browser/view/PRJEB36947,仅仅是提供bam文件,如果你搞不定表达矩阵,可以发邮件找我索取,然后完成学徒作业!!!
历年学徒作业目录如下:
- 生信编程直播课程优秀学员作业展示1
- 生信编程直播课程优秀学员学习心得及作业展示3
- 生信编程直播课程优秀学员作业展示2
- 给学徒的GEO作业
- 这个WGCNA作业终于有学徒完成了!
- 上次说的gmt函数(学徒作业)
- 拖后腿学徒居然也完成作业,理解RNA-seq数据分析结果
- 肿瘤外显子视频课程小作业
- ChIPseq视频课程小作业
- Agilent芯片表达矩阵处理(学徒作业)
- 学徒作业:TCGA数据库单基因gsea之COAD-READ
- 学徒作业-在CCLE数据库里面根据指定基因在指定细胞系里面提取表达矩阵
- 学徒作业-指定基因在指定组织里面的表达量热图
- 学徒作业-我想看为什么这几个基因的表达量相关性非常高
- 学徒作业:给你8个甲基化探针, 你在tcga数据库进行任意探索
- 学徒作业-根据我的甲基化视频教程来完成2015-NPC-methy-GSE52068研究
- RNA芯片和测序技术的比较(学徒作业)
- 学徒作业-单基因的tcga数据挖掘分析
- ATCC终于出来了organoids资源
- 拿到7个DDR通路的基因集-学徒作业
- 绘图本身很简单但是获取数据很难
- 都说lncRNA只有部分具有polyA尾结构,请证明
- 学徒作业-hisat2+stringtie+ballgown流程
- 学徒任务-探索DNA甲基化的组织特异性
- 用WES和RNA-Seq数据提取到的somatic SNVs不一致
- 《GEO数据挖掘课程》配套练习题