翻译自: https://constantamateur.github.io/2020-06-09-scBatch1/
更多教程见其博客:https://constantamateur.github.io/
Part 1- 从UMAP观察批次效应 2020/6/9
随着单细胞转录组数据集的不断增加,人们越来越频繁地对数据进行联合分析。
当然,这样做的第一件问题就是数据来自不同样本,实验室和实验的数据并不能“很好地混合”。数据不能很好的联合分析主要是基于细胞注释和使用UMAP 和/或 tSNE 进行的降维分析来检查数据是否能继续分析。通常是应用某种批次矫正方法来去除批次效应。
能提供最多信息量的 scRNA-seq 仍是新鲜的组织实验,需要新鲜的组织。这一要求阻碍了平衡实验设计,因为大多数样本在组织可用时都是按顺序处理的。因此,即使在同一实验室使用相同技术进行的封闭实验中,也会出现批次效应的问题。
在一系列帖子的第一篇中,作者说过,在这个领域中大家一般都会对批次效应进行过度矫正。这并不是说永远不应该使用批次矫正。作者自己也参与了一种单细胞批次矫正 method。但是他认为,急于纠正一切看起来像是批次效应的事情,有可能会掩盖了有趣的生物学,也许背后藏着潜在的生物学意义。
我们的公众号生信技能树之前也写了很多关于批次效应的教程,例如
大家可以当做是背景知识读一下!
判断是否是批次效应
假设我们有两个样本,并且对两个样本在同一组织上进行了10X 仪器的 3’基因检测。
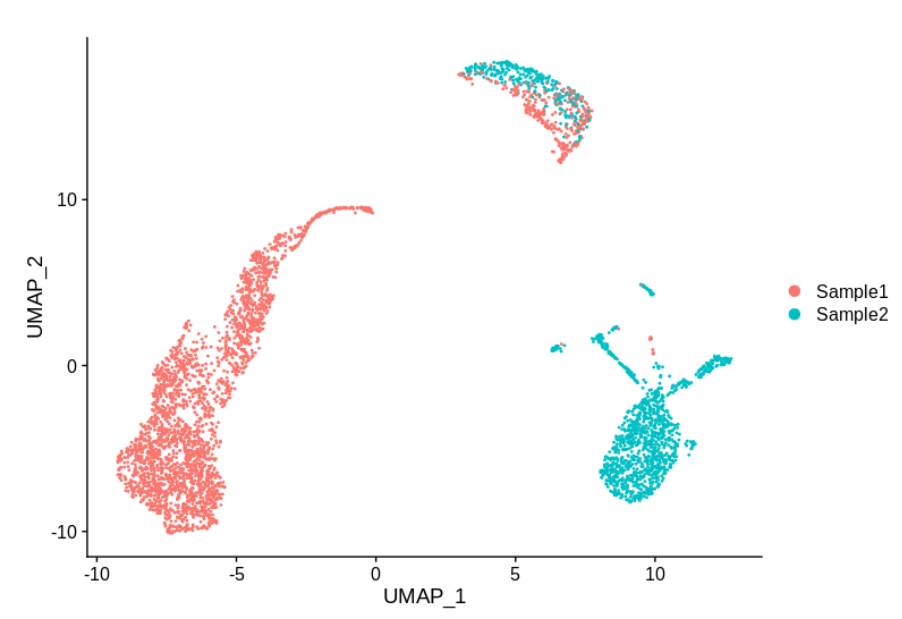
这个例子完全是虚构的 (虽然使用的是真实数据),但作者见过大概类似的图(可能作者是在说这不是随便瞎编的)。那么,这个数据有没有需要矫正的批次效应呢?顶部的细胞簇合理,但左侧和右侧的组特定于每个样品。那么应该进行批次矫正吗?
是否应该使用批次矫正,如果是的话,应该使用方法X,Y,或Z。实际上这不是一个实际的问题。作者在问一些更基本的问题。这里有没有批次效应需要担心并使用一些工具来矫正这些批次效应?有些人可能会争辩说,当不存在批次效应的时候,对数据进行合理的批次效应矫正不会对数据产生影响或者仅仅是一点点影响。但没有一种方法是完美的,我们不应该对数据做不必要的处理。
在我看来,正确的答案是我们没有足够的信息来判断此处是否存在批次效应。要做出决定,我们确实需要对每个簇所代表的细胞类型/状态有所了解。让我们考虑两种可能的注释。
注释A:
假如我们把上面的两个样品进行聚类分群后,发现它们属于下面的不同的细胞亚群注释;
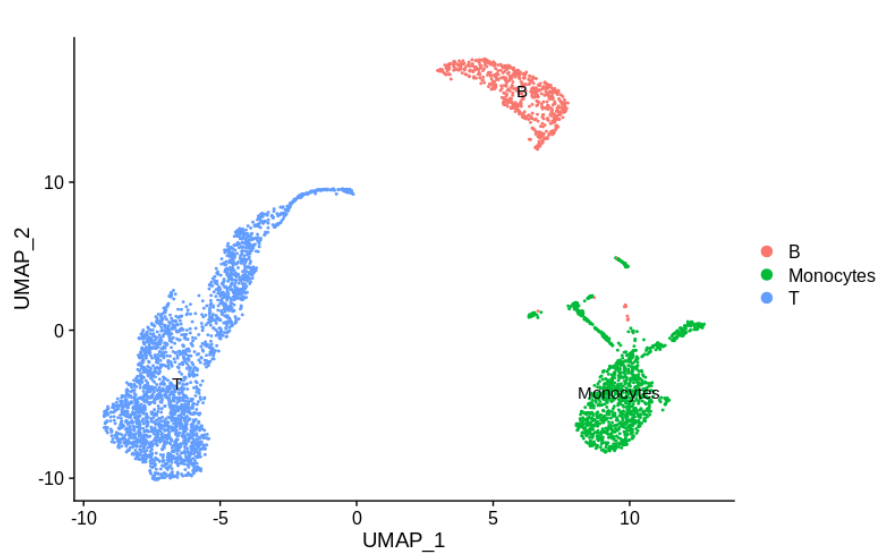
这里样本特定的簇代表不同的细胞类型,单核细胞和T细胞,而合理混合的簇代表B细胞。
显然,我们想研究为什么样本 1 中没有单核细胞,而样本 2 中没有T细胞。
但是,我们当然不希望尝试使用批次矫正来使单核细胞看起来像T细胞。
实际上,任何明智的单细胞批次矫正方法都不会去合并这些簇,只会尽量使它们分开。
所以,这样的注释情况下,是无需进行批次效应的去除!
注释 B:
假如上面的细胞亚群如下所示:
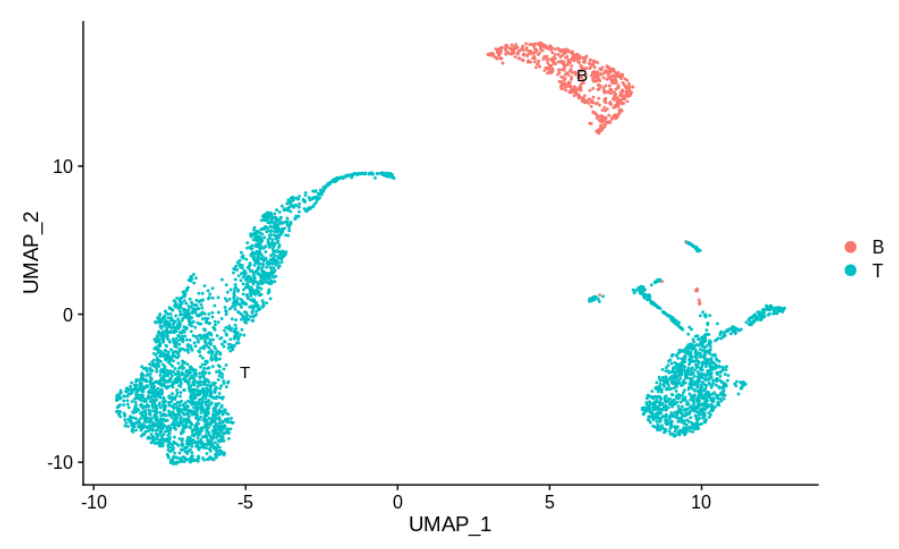
这里T细胞分为两组,每组特定于一个样本。
作者认为大多数人会很乐意声称这是批次效应的良好证据,并尝试进行矫正。就个人而言,在决定之前,仍然需要更多信息。其中一批细胞在批次之间(B细胞)混合得很好。
尽管技术批次效应可能是非线性的,并以不同的方式影响不同的簇,但大家仍然想确切地知道两个T细胞簇之间的差异。
作者的观点是,发现批次效应和矫正批次效应从根本上说是生物学的,而不是技术的。
确定存在批次效应,是由于上图中的差异超出了我们预期的T细胞组成或生物学差异。或者我们根本不在乎这种变化,而只对批次之间的相似性感兴趣。
到底有什么不同
需要多大的差异才能促使在 UMAP / tSNE 图形中聚集为不同的簇?
作者的经验:基本上通过查看点在数据的 2D 呈现出来的聚类就可以判断此单细胞数据中是否有批次效应。你是否真的知道两组细胞有多大的差异才会在 UMAP 图上形成两个簇?
我对 UMAP (以及像 Louvain 这样的基于图形的聚类方法)背后的数学理解不够深入,无法预测转录组需要多少差异才能在 UMAP 图上显示为不同的簇。所以下面用 10X PBMC data 数据做了一些实验。加载数据并在Seurat中进行了如下处理,
library(Seurat)
quickCluster = function(dat,numPCs=75,clusteringRes=1.0){
ss = CreateSeuratObject(dat)
ss = NormalizeData(ss,verbose=FALSE)
ss = FindVariableFeatures(ss,verbose=FALSE)
ss = ScaleData(ss,verbose=FALSE)
ss = RunPCA(ss,npcs=numPCs,approx=FALSE,verbose=FALSE)
ss = FindNeighbors(ss,dims=seq(numPCs),verbose=FALSE)
ss = FindClusters(ss,res=clusteringRes,verbose=FALSE)
ss = RunUMAP(ss,dims=seq(numPCs),verbose=FALSE)
return(ss)
}
#Start with PBMC data
pbmc = Read10X('PBMC_4k/filtered_gene_bc_matrices/GRCh38/')
pbmc = quickCluster(pbmc)
DimPlot(pbmc,label=TRUE)
这个作者自定义的 quickCluster 函数非常有意思,可以最快的速度看看我们的单细胞转录组表达矩阵的一般特性!
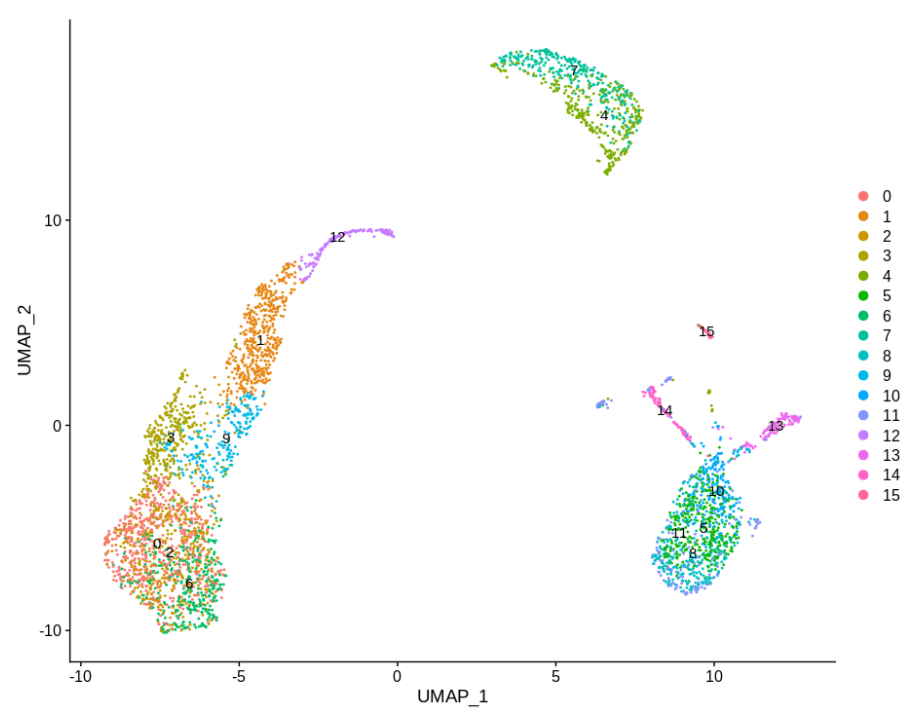
取出 cluster 4和 cluster 7,因为它们相当均匀,并以此为实验基础。
w = which(pbmc@active.ident %in% c(4,7))
bb = pbmc@assays$RNA@counts[,w]
bb = quickCluster(bb) ## 整理重新走一波单细胞转录组标准分析流程哦,调用了前面定义好的quickCluster函数
还是上面那个问题,需要多大的差异来促使 UMAP 中分为不同的簇,将这一细胞集分为两半,并作为两个“批次”。
然后需要改变多少个基因才能在这些批次中诱导不同的肉眼可见的明显分离开的簇。下面改变1、2、5、10和20个基因的结果。
#' Alter genes in a random subset of cells
#'
#' @param srat Seurat object.
#' @param genes A vector of genes that will be altered.
#' @param tgtVals Values to set genes to. Defaults to zero.
#' @param frac Fraction to modify genes for.
#' @return Altered Seurat object.
alterCellSubset = function(srat,genes,tgtVals = rep(0,length(genes)),frac=0.5){
# Get the random subset
w = sample(nrow(srat@meta.data),ceiling(nrow(srat@meta.data)*frac))
mtx = srat@assays$RNA@counts
#Alter matrix
mtx[genes,w] = tgtVals
#Do clustering
srat = quickCluster(mtx)
srat@meta.data$modified = seq(ncol(mtx)) %in% w
srat@meta.data$batch = ifelse(srat@meta.data$modified,'Batch1','Batch2')
return(srat)
}
gns = rownames(bb@assays$RNA@counts)[order(Matrix::rowSums(bb@assays$RNA@counts>0))]
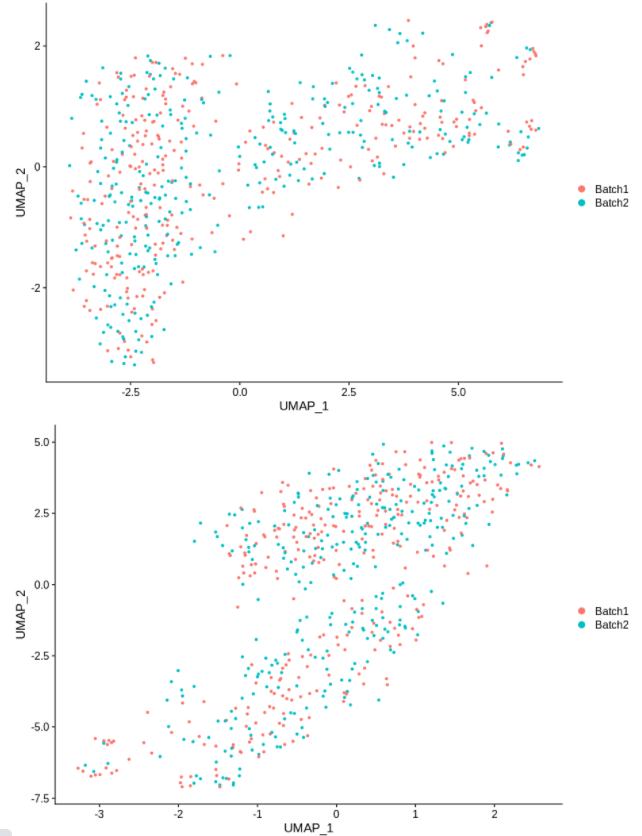
cc1 = alterCellSubset(bb,tail(gns,1))
DimPlot(cc1,group.by='batch')
cc2 = alterCellSubset(bb,tail(gns,2))
DimPlot(cc2,group.by='batch')
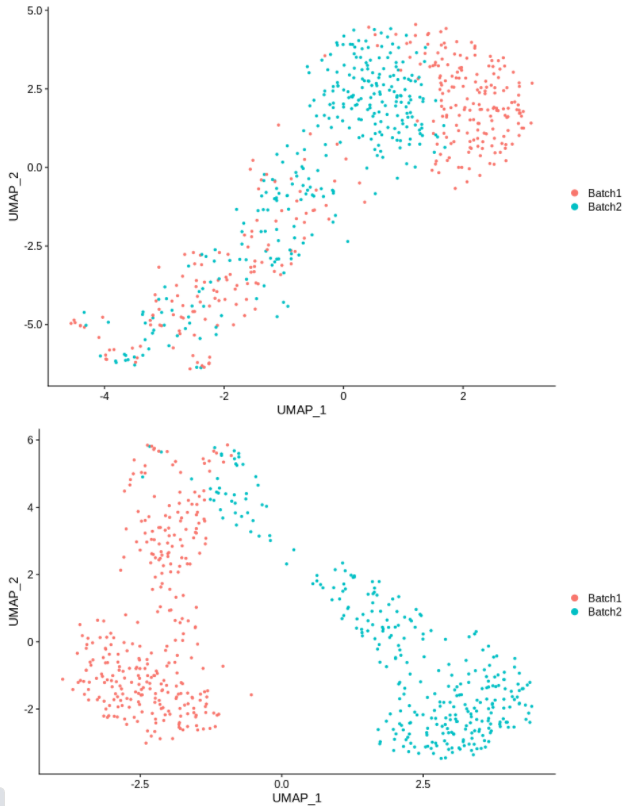
cc5 = alterCellSubset(bb,tail(gns,5))
DimPlot(cc5,group.by='batch')
cc10 = alterCellSubset(bb,tail(gns,10))
DimPlot(cc10,group.by='batch')
cc20 = alterCellSubset(bb,tail(gns,20))
DimPlot(cc20,group.by='batch')
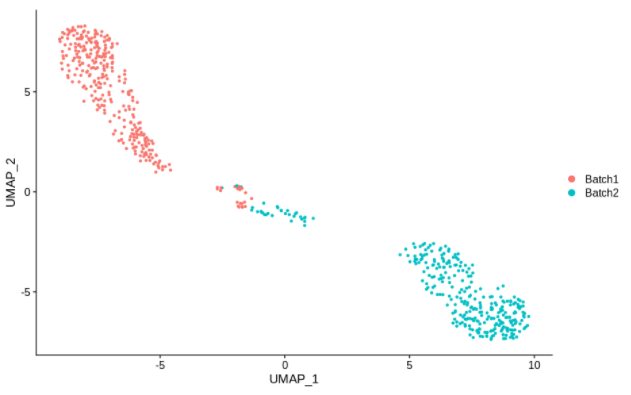
画上面这些图时,作者选择了最有可能在敲除时产生影响的基因。
尽管如此,令人震惊的是,在一组表达15000个不同基因的细胞中,仅仅改变其中的5个就足以带来如此大的变化。
仅仅改变2个基因时,都有一些模糊的迹象表明簇即将会分离。计算一下批次的混合熵,并绘制了随着基因变化而变化的函数,
#' Calculates the batch mixing entropy for a Seurat object with meta.data 'batch'
#'
#' @param srat Seurat object. Must have NN calculated and columns "modified" and "batch" in the meta-data.
#' @return The batch entropy.
calcEntropy = function(srat){
tgtCells = rownames(srat@meta.data)[srat@meta.data$modified]
cNgbs = as.matrix(srat@graphs$RNA_nn[tgtCells,]>0)
ent = 0
batch = srat@meta.data$batch
kk=0
for(tgtCell in tgtCells){
kk = kk + 1
lCnts = table(batch[which(cNgbs[kk,])])
lFreq = lCnts/sum(lCnts)
ent = ent -sum(lFreq*log(lFreq))
}
return(ent)
}
#Make plot of entropy by killed gene
nDropped = seq(0,20)
ents = lapply(nDropped,function(e) calcEntropy(alterCellSubset(bb,tail(gns,e))))
plot(nDropped,unlist(ents),
type='l',
frame.plot=FALSE,
xlab='# Genes removed',
ylab='Batch entropy'
)
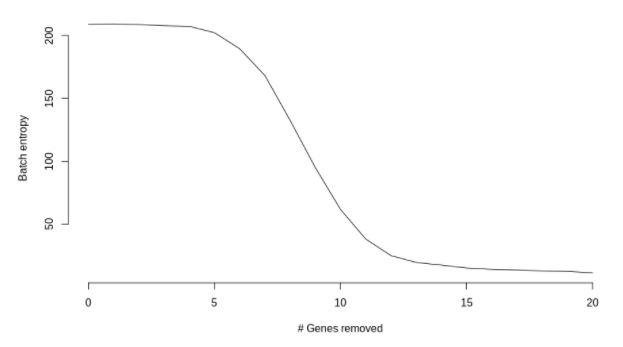
随着修饰基因数量的增加,这几乎呈现出单调的趋势,趋向于更低的熵(批次之间的混合更少)。一旦两组细胞基本完全分离,熵的变化就会减慢。
这个结果并没有令人很惊讶。使用复杂降维技术(如 UMAP )的全部意义在于,应该能够检测出细微的差异。
这不是 UMAP 的缺点,这正是这类算法所需要的。但它能真正告诉我们的是,这些细胞群之间是有差异的。这取决于详细地研究它们,并决定这些差异是否有意义。
那么我们应该做些什么呢?作者希望很快就能写第二篇关于这个问题的帖子。(其实作者已经写完了,但是我们的翻译工作滞后,请大家看明天生信技能树的教程哈!)
但至少,作者认为我们应该认识到,批次效应的判断和矫正是由于我们需要背后的生物学意义(我自己猜测这句话的意思是,不是每个数据一上来有点差异就开始矫正,而是需要仔细思考一下是否需要)。
批次效应的判断是基于降维曲线进行的,那么少量基因就可以促使批次效应。
考虑到这一点,花时间找出哪些基因使批次分离出不同的簇(这并不需要花费太多的时间),难道不值得吗。这样做可以揭示促使这些簇分开的技术效应的本质,或者也许可以发现有趣的生物差异,否则这些差异就会被忽略。