昨天提到了最近接了一个单细胞转录组项目,有80个10X样品,每个样品的单细胞测序数据都是100G左右的fq.gz文件,在跑完了cellranger流程后整理结果的同时,重新捡起来了七八年前的Linux知识,写了个 带着文件夹结构的拷贝笔记分享给大家。
其实这个项目背后的故事还有很多,我安排给一个萌新负责跑这个流程。因为确实超级简单,我写了一个脚本,文件名是 run-cellranger.sh ,内容如下所示 :
bin=../pipeline/cellranger-5.0.1/bin/cellranger
db=../pipeline/refdata-gex-GRCh38-2020-A
ls $bin; ls $db
fq_dir=/home/data/project/10x/raw
$bin count --id=$1 \
--localcores=4 \
--transcriptome=$db \
--fastqs=$fq_dir \
--sample=$1 \
--expect-cells=5000
然后每个样本都只需要提交上面的这个脚本即可,示例如下 :
bash run-cellranger.sh YX-Endo-Decidu 1>log-YX-Endo-Decidu.txt 2>&1
# 每个样品都是独立的提交方式
但是,那萌新也太让我不省心了,不知道他在哪学的Linux脚本编写方式,把全部的80个样品的命令打印出来成为一个脚本,然后nohup那个脚本。
我等了两天后询问项目进展,结果跟我说才跑了8个样品,我勒个去,这是一个加急项目啊!所以我过去给他纠正了,让他学会并行处理项目,不仅仅是批量处理。
虽然我每个10x样品里面的代码都是调用了4个线程,但是样本很多,这个时候把多个样本同时提交,也就是并行,理论上也可以加快这个项目进度,当然了,前提是这个服务器有足够的计算资源,都可以给这个项目调配。
然后另外一个更气人的事情就出现了,该萌新居然是把80多个10x样本的run-cellranger.sh 同时全部提交了!!!
然后我们的服务器就崩溃了,唉,如下所示:
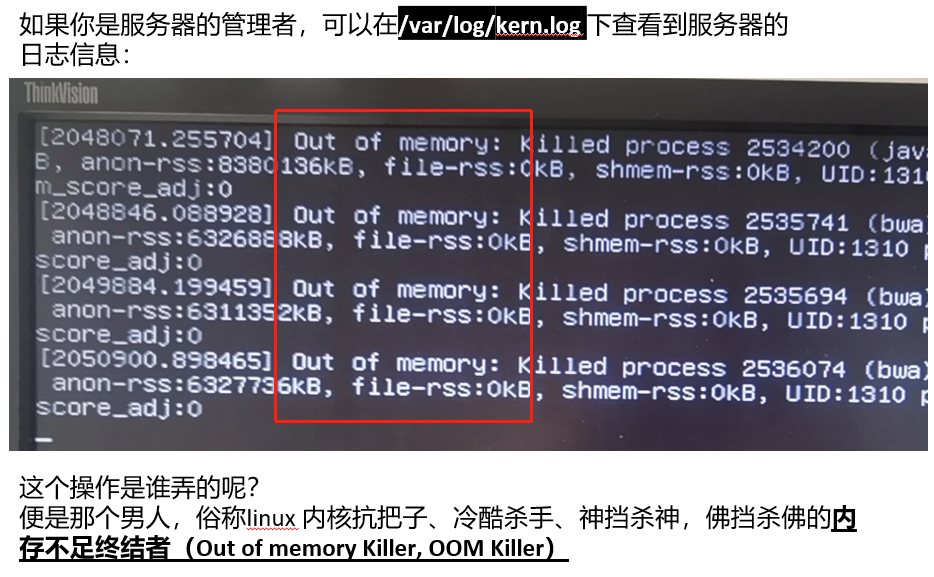
因为找不到真正的cellranger把服务器搞奔溃的截图,所以只好是放了一个全面实习生的“血的教学”。其实绝大部分生物信息学的软件工具都是设置了内存和CPU接口,不可能让大家无限制消耗计算机资源的,我让实习生总结了,如下:
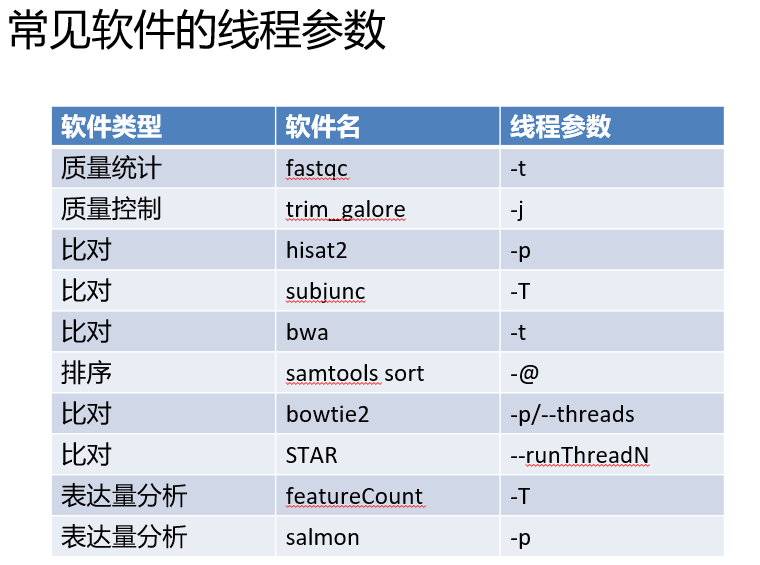
我们的服务器目前并没有组建集群,我拿出来了其中一个96线程372G内存的单机给这个80多个10x样本数据处理项目,其实稍微计算一下就明白,应该是每次提交20个样品的run-cellranger.sh 脚本,分成4次提交即可。
这个时候,需要使用下面的一些shell语法啦,主要就是每次运行样品,都需要对一个计数变量自增 1
config_file=$1
number1=$2
number2=$3
index=/reference/index/salmon/gencode.v32.salmon.index
mkdir -p quants
cat $config_file |while read id
do
file=$(basename $id )
sample=${file%%.*}
if((i%$number1==$number2))
then
arr=($id)
fq1=${arr[1]}
fq2=${arr[2]}
sample=${arr[0]}
echo "Processin sample ${sample}"
salmon quant -l IU --gcBias -i $index -1 $fq1 -2 $fq2 -p 4 -o quants/${sample}_quant
fi
i=$((i+1))
done
我实在是不明白,这个脚本很难吗?
我在R里面给萌新写了个例子;
f=1:24
f
echo <- function(f,n1,n2){
n=1
for (i in f ){
if ((n %% n1) == n2) {
print(i)
}
n=n+1
}
}
echo(f,4,1)
确实很简单啊?
编程能力其实是解决问题的能力