前面我们公布了《cytof数据资源介绍(文末有交流群)》,现在就开始正式手把手教学。
上一讲我们构造好了SingleCellExperiment对象,后续全部的分析都会以这个SingleCellExperiment对象为准,大家务必熟悉SingleCellExperiment对象的各种结构,教程见:cytofWorkflow之构建SingleCellExperiment对象(二)。有了这个SingleCellExperiment对象,而且经过了合理的质量控制,并且聚类分群了,就可以开始整理它亚群并且根据抗体信号强度值对不同亚群进行生物学注释。
前面的教程:cytofWorkflow之聚类分群(四),每个病人的20个亚群细胞都区分出来了。但是这20群是 人为指定的,可以放大或者缩小。
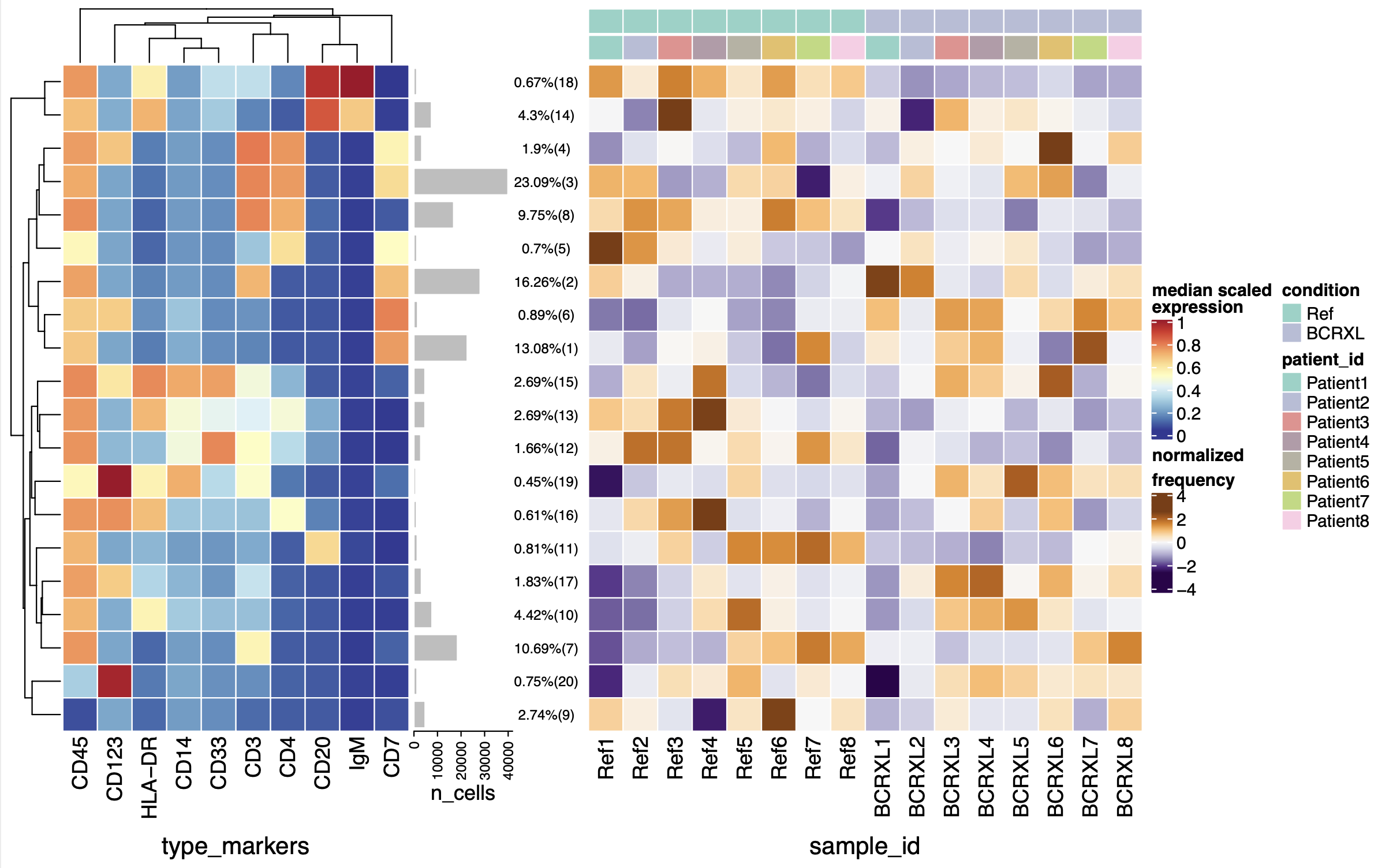
首先可以查看每个抗体的信号强度情况
代码如下:
for (gene in names(sce@rowRanges)) {
plotDR(sce, "TSNE", color_by = gene )
ggsave2(filename = paste0(gene,'_gene_tSNE.pdf'))
}
可以看到有一些抗体是互补的,比如 CD3 CD45 CD4 ,它们就是主要的分群标志物。
自动注释
目前我做cytof数据的细胞亚群的生物学自动注释的包很少,有下面的两个还在预印本:
当然了,可以搜索到更多,我就不代劳了,有一个Bioinformatics. 2017 Jun 的文章《Automated cell type discovery and classification through knowledge transfer》提出来了一个python包可以做Mass cytometry by time-of-flight (CyTOF) 的细胞分群和注释。
我这里想演示的自动注释,其实仍然是手动的代码,只不过呢,这个代码,在很多数据集是可以共用的。
首先使用下面的代码,查看不同的抗体在不同的细胞亚群(总共20个)的信号强度矩阵:
rm(list = ls())
suppressPackageStartupMessages(library(flowCore))
suppressPackageStartupMessages(library(diffcyt))
suppressPackageStartupMessages(library(HDCytoData))
require(cytofWorkflow)
load(file = 'K20_output_of_cytofWorkflow.Rdata')
sce@metadata
p=plotExprHeatmap(sce, features = "type",
by = "cluster_id", k = "meta20",
row_clust = F,
bars = TRUE, perc = TRUE)
p
如下所示:
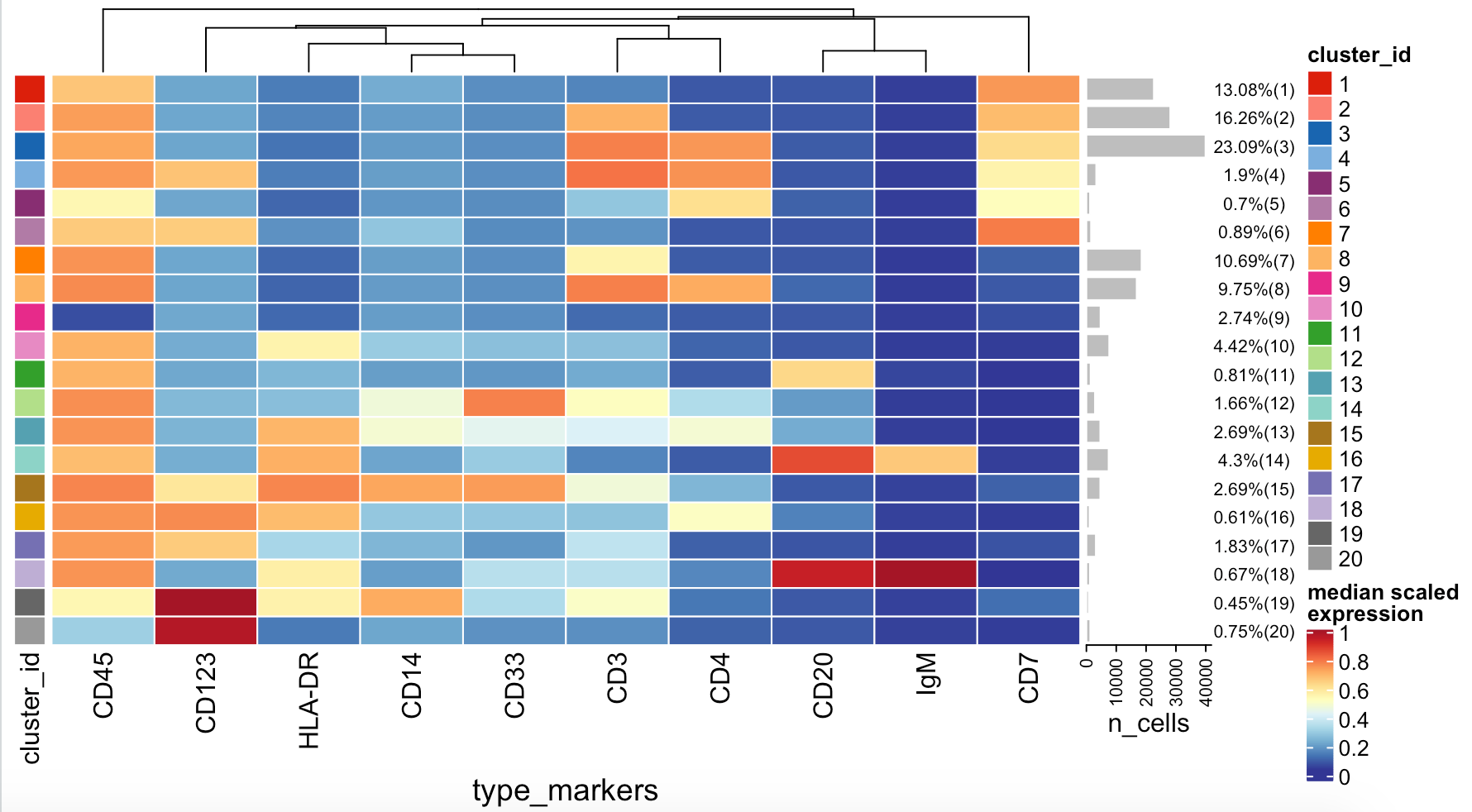
既然有了这20个细胞亚群的不同抗体信号强度情况,就可以逐步分类,下面是一个示范代码:
p@matrix
kp=p@matrix > 0.5
colnames(kp)
# "CD45" "F4/80" "CD19" "CD3" "CD4"
k1=which(kp[,"CD45"] & kp[,"CD3"] & kp[,"CD4"] );k1 # CD4
k2=which(kp[,"CD45"] & kp[,"CD3"] & !kp[,"CD4"] );k2 # CD8
k3=which(kp[,"CD45"] & kp[,"CD20"] & !kp[,"CD3"] );k3 # Bcells
k4=which(kp[,"CD45"] & kp[,"CD123"] & !kp[,"CD3"] & !kp[,"CD7"] & !kp[,"CD14"] );k4 # DC
k5=which(kp[,"CD45"] & kp[,"CD14"] );k5 # monocytes
k6=setdiff(1:20,c(k1,k2,k3,k4,k5));k6
merging_table1=data.frame(id=c(k1,k2,k3,k4,k5,k6),
new_cluster=c(rep("CD4",length(k1)),
rep("CD8",length(k2)),
rep("Bcells",length(k3)),
rep("DC",length(k4)),
rep("monocytes",length(k5)),
rep("other",length(k6))) )
merging_table1
上面的代码仅仅是一个演示而已,具体如何分群,完全是取决于生物学背景知识啦,请不要照抄上面的代码。
手工注释
需要注意的是,我们这个时候,虽然使用的是同样的数据,但是呢,我们的群的顺序变化了哈,所以呢,没办法直接使用作者的分群命名方式:
如果一切按照教程来,是可以直接下载作者的生物学分群命名的:
library(readxl)
url <- "http://imlspenticton.uzh.ch/robinson_lab/cytofWorkflow"
merging_table1 <- "PBMC8_cluster_merging1.xlsx"
download.file(file.path(url, merging_table1),
destfile = merging_table1, mode = "wb")
merging_table1 <- read_excel(merging_table1)
head(data.frame(merging_table1))
可以看到作者定义的生物学群是:
original_cluster new_cluster
1 B-cells IgM+
2 surface-
3 NK cells
4 CD8 T-cells
5 B-cells IgM-
6 monocytes
7 monocytes
8 CD8 T-cells
9 CD8 T-cells
10 monocytes
11 monocytes
12 CD4 T-cells
13 DC
14 CD8 T-cells
15 CD4 T-cells
16 DC
17 CD4 T-cells
18 CD4 T-cells
19 CD4 T-cells
20 CD4 T-cells
原作者的结果是;
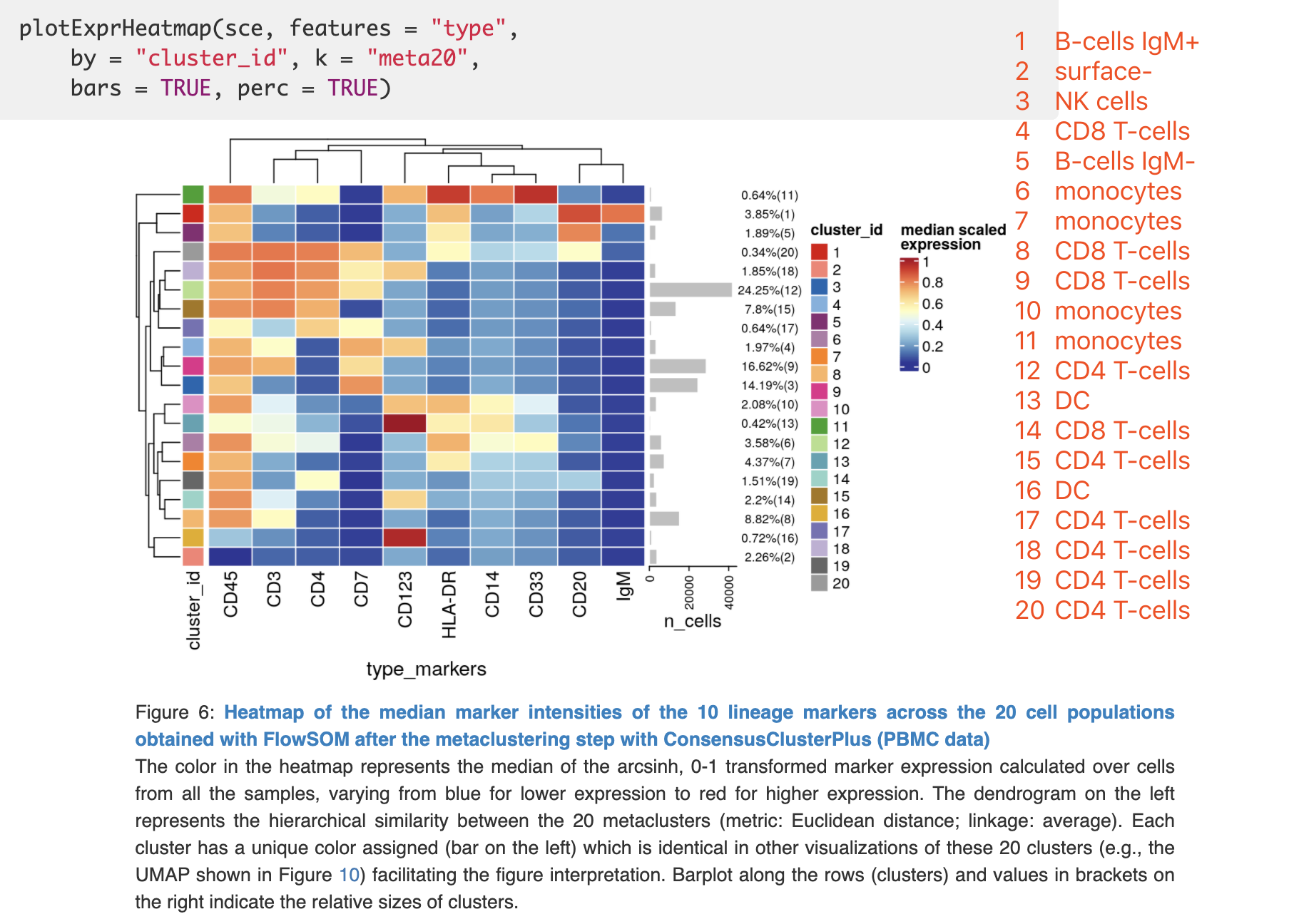
基本上是根据生物学意义来的,CD4和CD8的T细胞都是高表达CD45和CD3,然后分别表达CD4和CD8。表达CD14的就归类为monocytes,超高表达CD123的认为是DC细胞亚群,表达CD20的是B细胞,然后B细胞有2个亚群,所以命名为B-cells IgM+和B-cells IgM- 即可。
非常容易理解, 但前提是你有这样的生物学背景知识。之所以归类为手工注释,就是说,可以直接把上面的热图结果丢给你的项目合作者,让他们首先命名后给你表格,然后你就可以走后面的代码进行合并啦!
合并亚群
前面我们把20个亚群,都根据生物学背景重新命名后,就可以合并啦,代码如下:
colnames(merging_table1)=c('original_cluster' , 'new_cluster')
length(unique(merging_table1$new_cluster))
# apply manual merging
sce <- mergeClusters(sce, k = "meta20",
table = merging_table1, id = "merging1",overwrite = TRUE)
library(ggsci)
n=length(unique(merging_table1$new_cluster))
cl=pal_lancet("lanonc",alpha = 0.6)(n)
cl
plotDR(sce, "TSNE",
k_pal=cl,
color_by = "merging1")
这个时候呢,就需要考虑配色的问题,所以我前面使用 了 ggsci包,出图如下:
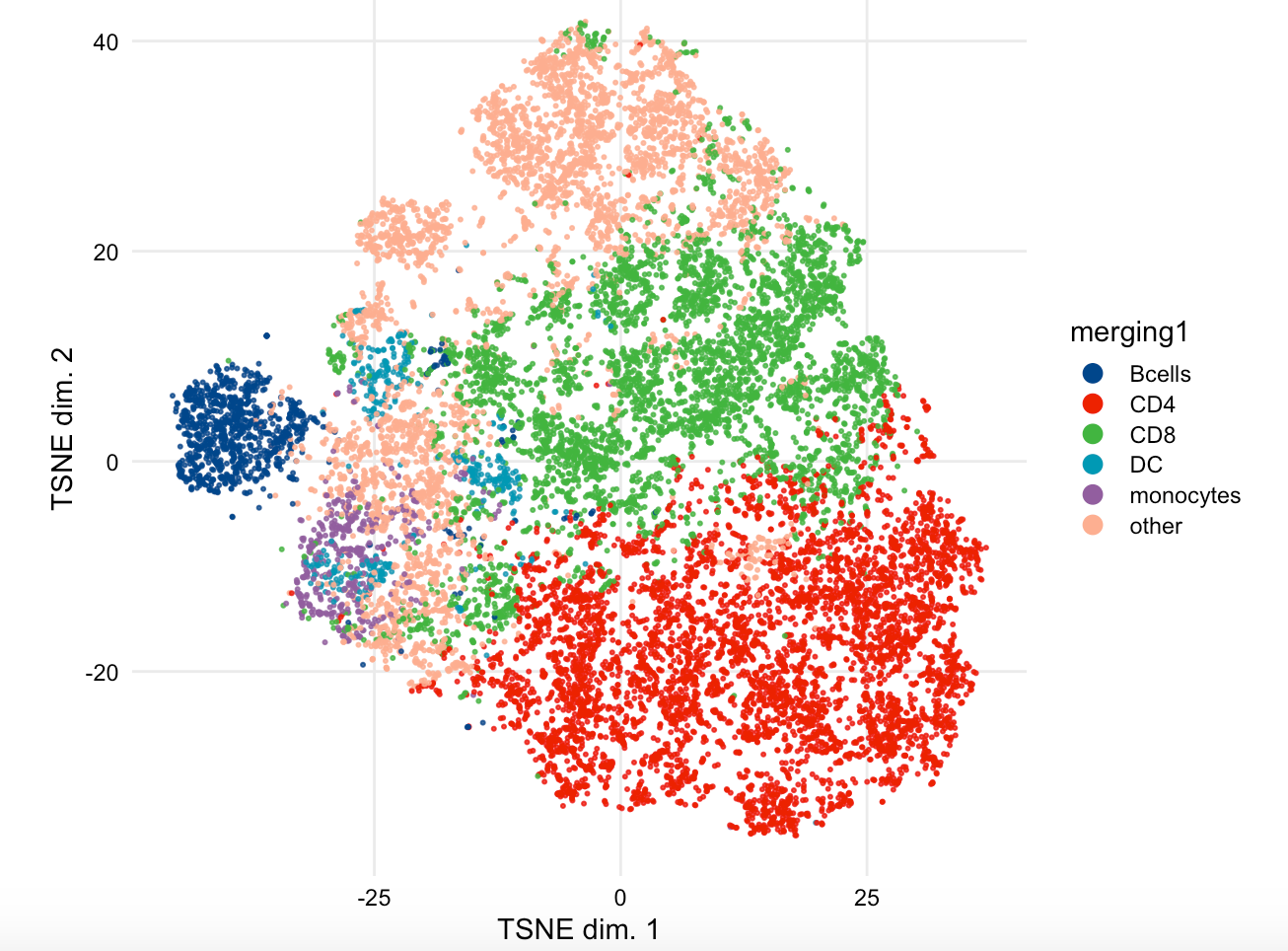
可以看到, 细胞分群后,重新命名并不是很合理,因为生物学背景不够。