在朋友圈刷到了这个文章,2020年1月4日,中国医学科学院北京协和医学院朱兰及中国科学院北京基因组研究所杨运桂共同通讯在Nature Communications 在线发表题为“Single-cell transcriptome profiling of the vaginal wall in women with severe anterior vaginal prolapse”的研究论文,链接是:https://www.nature.com/articles/s41467-020-20358-y
我注意到这个研究比较好的结合了传统bulk转录组数据和单细胞转录组数据,值得解读和推荐给大家。
传统bulk转录组数据
链接是: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151188
15个样本:
GSM4568149 N1_RNA-seq
GSM4568150 N3_RNA-seq
GSM4568151 P2_RNA-seq
GSM4568152 P3_RNA-seq
GSM4568153 P4_RNA-seq
GSM4568154 P5_RNA-seq
GSM4568155 P6_RNA-seq
GSM4568156 P7_RNA-seq
GSM4568157 P8_RNA-seq
GSM4568158 P9_RNA-seq
GSM4568159 P10_RNA-seq
GSM4568160 P11_RNA-seq
GSM4568161 P12_RNA-seq
GSM4568162 P13_RNA-seq
GSM4568163 P14_RNA-seq
上游流程是:
- FastQC (version 0.11.5).
- HISAT2 (version 2.0.5)
- human reference genome (GRCm37/hg19; Ensembl version 72).
- FeatureCounts (version 1.6.0)
下游流程是:
- edgeR R package (version 3.18.1)
- |Log2-fold change | >0.5 and p-value < 0.05 as thresholds.
这些知识点, 我在B站有免费教学视频,观看方式 如下:
-
视频免费在B站:https://www.bilibili.com/video/BV12s41137HY 大家学习的时候记得发弹幕交流哈。
-
也有微云离线版本视频下载本地播放:
-
- 上游分析视频以及代码资料在:https://share.weiyun.com/5QwKGxi
- 下游主要是基于counts矩阵的标准分析的代码 https://share.weiyun.com/50hfuLi
-
同步查看视频配套代码 :https://www.jianshu.com/p/a84cd44bac67
-
RNA-SEQ实战演练的素材:https://share.weiyun.com/5h1Z2QY ,包括一些公司PPT,综述以及文献以及测试数据
-
RNA-SEQ 实战演练的思维导图:文档链接:https://mubu.com/doc/38y7pmgzLg 密码:p6fo
图表也很容易理解,就是差异分析的火山图和上下调基因独立的生物学数据库注释:
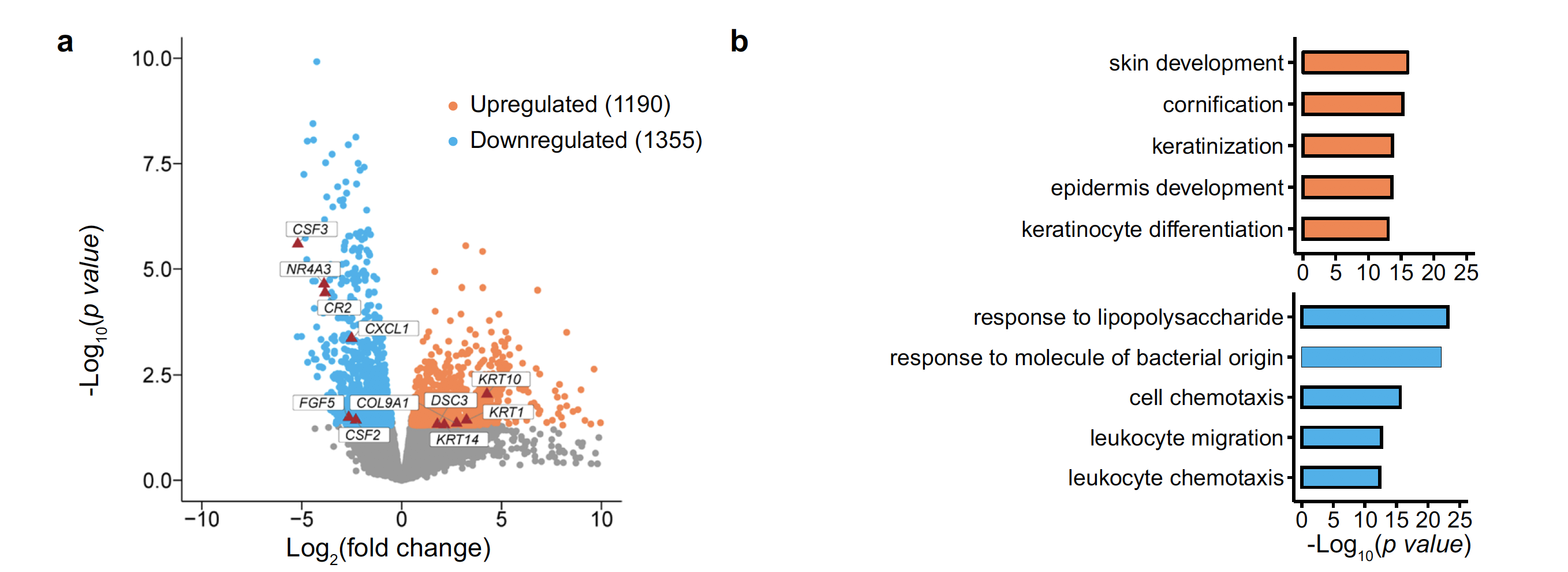
传统的转录组数据走差异分析,拿到了感兴趣的基因集后,通常是做超几何分布检验看看富集到了什么生物学功能数据库,比如KEGG或者GO数据库,或者走gsea/gsva这样的富集分析,也是注释生物学功能数据库。大家读我的表达芯片的公共数据库挖掘系列推文应该是够多了:
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
上图的基因集注释生物学功能数据库,就是下调基因是cell chemotaxis, leukocyte migration这样的功能,而上调基因是vaginal wall prolapse。因为是传统的bulk转录组数据,所以我们并不能确定这 1190 upregulated genes and 1355 downregulated genes 到底是由bulk转录组样品里面的具体什么细胞的改变造成的。
我们看到的其实是bulk转录组样品里面的全部细胞的改变的一个混合效应,但是如果加上单细胞转录组数据,就可以完美的解决这个问题:
- COL9A1, shown to be upregulated in bulk RNA-seq, was upregulated mainly in fibroblasts, whereas its expression was unchanged in other cells.
- Conversely, CXCL1 gene expression was downregulated in most cell types and upregulated in macrophages and mast cells.
- These differences reflect cellular heterogeneity in gene expression changes, further suggesting that investigating gene expression changes in each cell type in the prolapsed vaginal wall in POP is important.
前面的bulk转录组差异分析拿到的1190 upregulated genes and 1355 downregulated genes ,就被分解到了不同的细胞亚群。
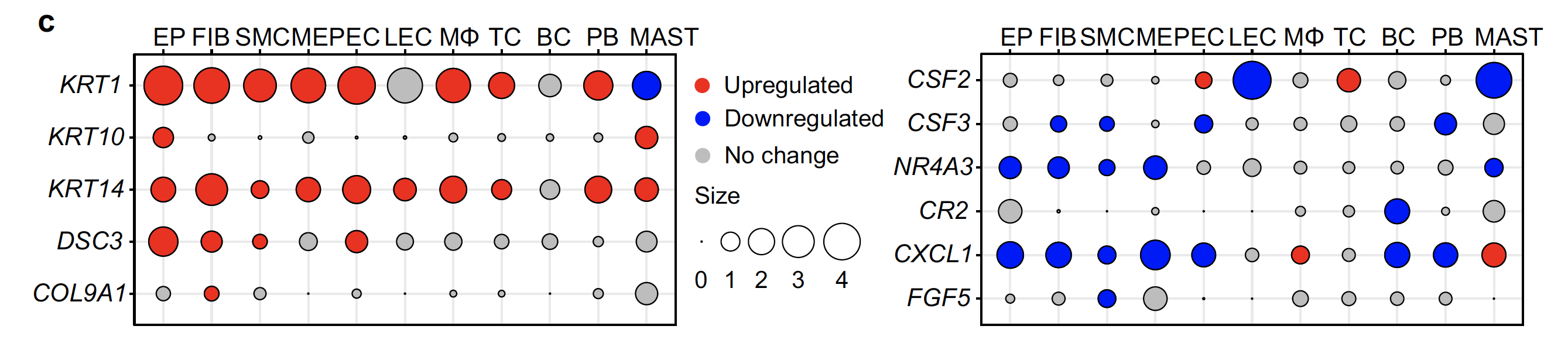
单细胞转录组(10X技术)
链接是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151192
GSM4568340 N1_scRNA-seq
GSM4568341 N2_scRNA-seq
GSM4568342 N3_scRNA-seq
GSM4568343 N4_scRNA-seq
GSM4568344 N5_scRNA-seq
GSM4568345 P1_scRNA-seq
GSM4568346 P2_scRNA-seq
GSM4568347 P3_scRNA-seq
GSM4568348 P4_scRNA-seq
GSM4568349 P5_scRNA-seq
GSM4568350 P6_scRNA-seq
GSM4568351 P7_scRNA-seq
GSM4568352 P8_scRNA-seq
GSM4568353 P9_scRNA-seq
GSM4568354 P10_scRNA-seq
GSM4568355 P11_scRNA-seq
GSM4568356 P12_scRNA-seq
GSM4568357 P13_scRNA-seq
GSM4568358 P14_scRNA-seq
GSM4568359 P15_scRNA-seq
GSM4568360 P16_scRNA-seq
这个就比较简单了,研究者也提供了全部的10X流程分析结果,大家可以在GSE151192数据集里面下载走Seurat流程,拿到下面的图表:
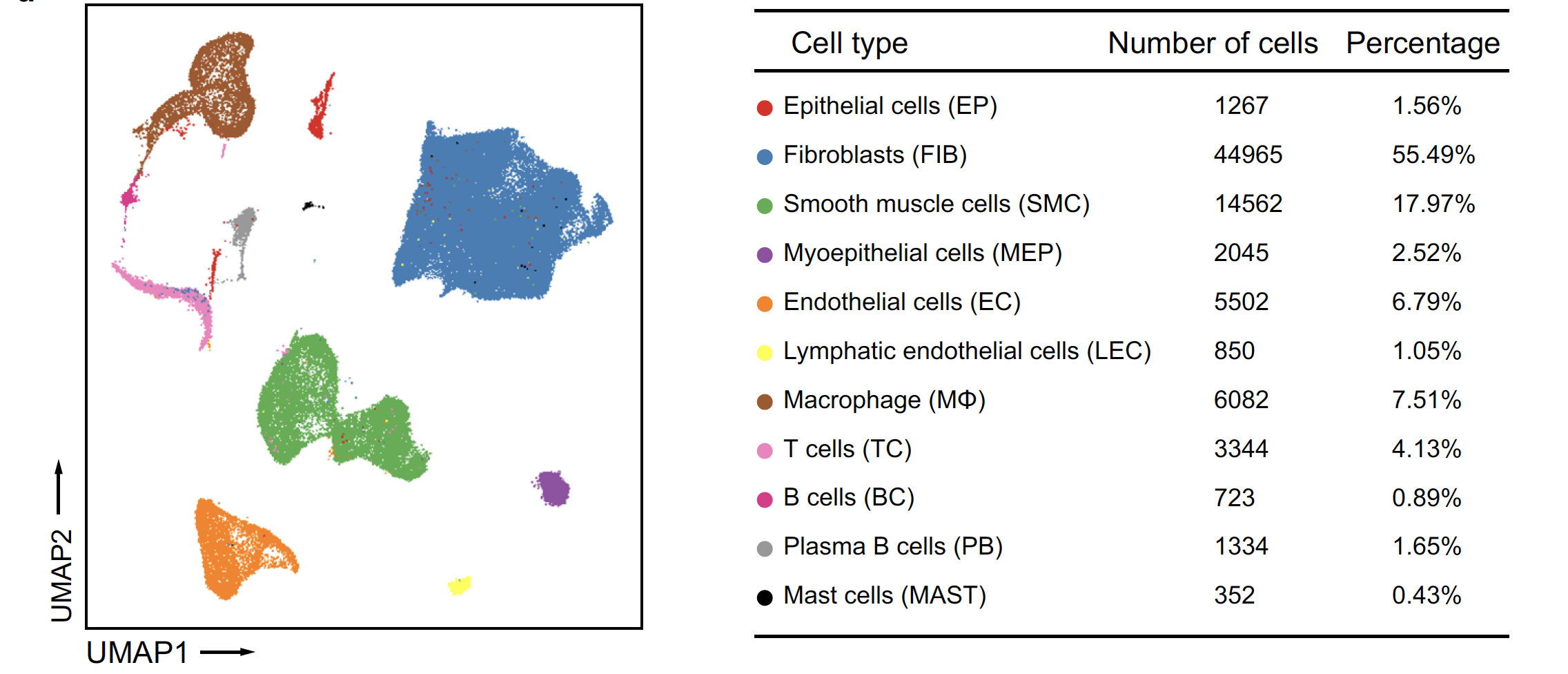
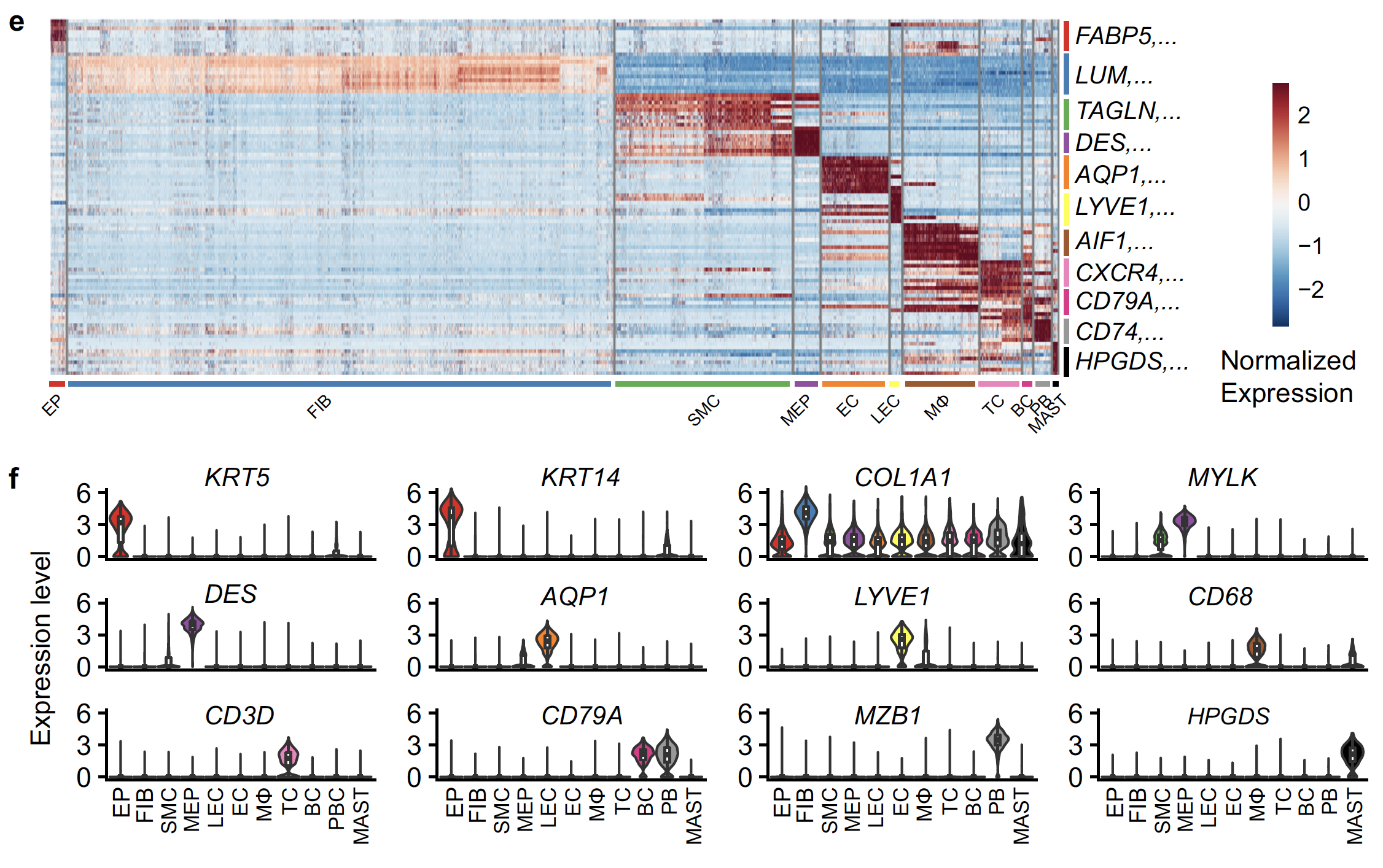
上面的质控降维聚类分群和细胞亚群的生物学注释比较简单,可以看看我们前面的例子:人人都能学会的单细胞聚类分群注释 。然后可视化不同细胞亚群的标记基因即可:
当然了,单细胞转录组数据分析肯定不仅仅是质控降维聚类分群和细胞亚群的生物学注释,以及可视化不同细胞亚群的标记基因这些标准分析啦。
后面还可以加上拟时序分析,转录因子分析,细胞通讯分析,这些都在“Single-cell transcriptome profiling of the vaginal wall in women with severe anterior vaginal prolapse”的研究论文有所体现,我们就不继续讲解了。去年我们在《生信技能树》公众号带领大家一起学习过:SCENIC转录因子分析结果的解读 ,以及:细胞通讯分析结果的解读。