三年多没有看我们《生信技能树》的论坛了,都差点忘记了这个产品,昨天不小心点击进去了,恰好看到了一个问题,就回复一下吧!
在:http://www.biotrainee.com/thread-8003-1-1.html 可以看到如下所示的提问:
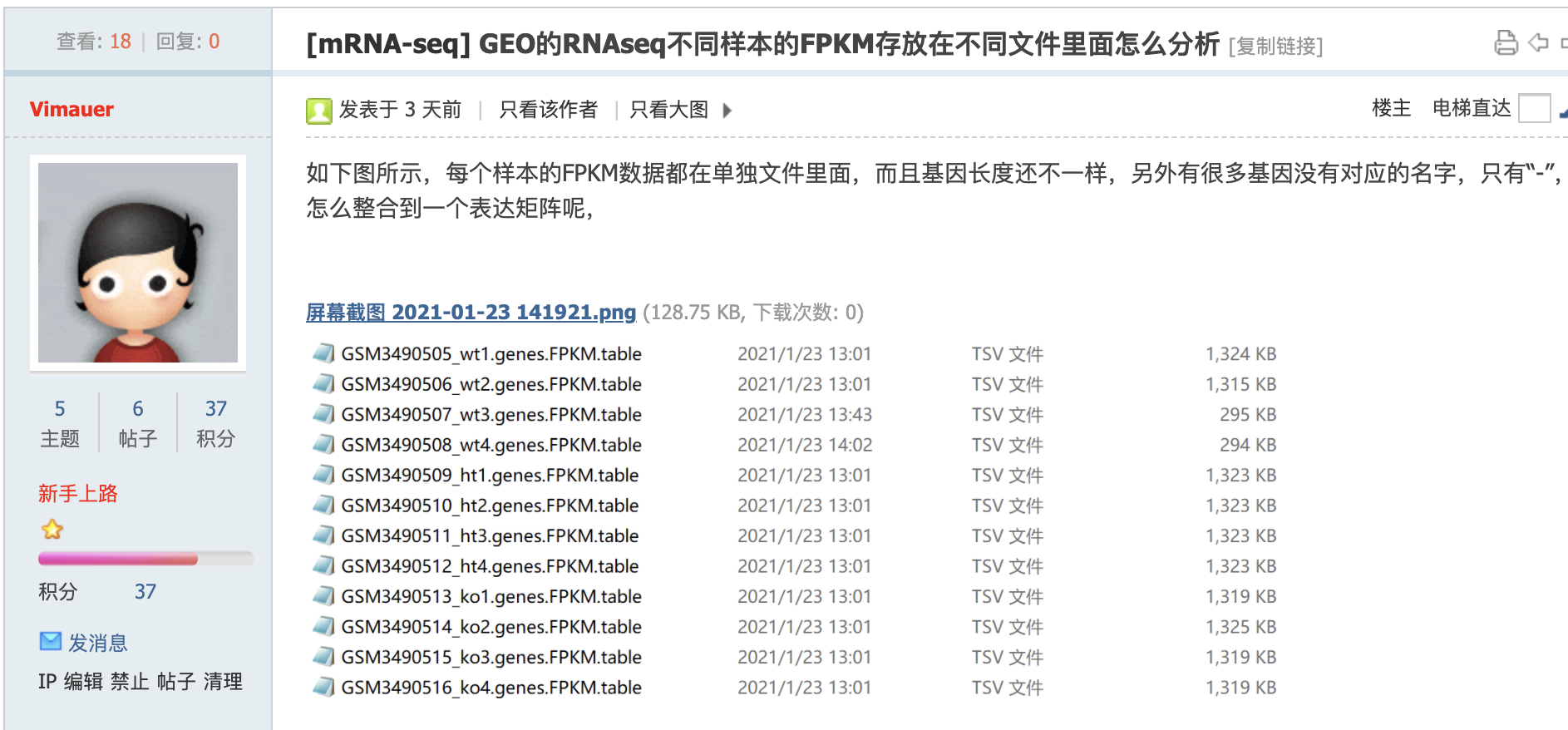
根据截图我找到了:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123005 数据集,其实这个读者问问题的时候,应该是自己描述清楚这个链接,避免浪费我的时间去猜测它的数据集。
这个数据集虽然很新,但是其测序仪有一点年代了,Illumina HiSeq 2500 (Mus musculus)
GSM3490505 RNA-seq_ApcWT-1
GSM3490506 RNA-seq_ApcWT-2
GSM3490507 RNA-seq_ApcWT-3
GSM3490508 RNA-seq_ApcWT-4
GSM3490509 RNA-seq_ApcHET-1
GSM3490510 RNA-seq_ApcHET-2
GSM3490511 RNA-seq_ApcHET-3
GSM3490512 RNA-seq_ApcHET-4
GSM3490513 RNA-seq_ApcKO-1
GSM3490514 RNA-seq_ApcKO-2
GSM3490515 RNA-seq_ApcKO-3
GSM3490516 RNA-seq_ApcKO-4
我的代码如下
首先是批量读取啦,如下:
fs=list.files('GSE123005_RAW/')
fs
dat=lapply(fs, function(i){
read.table(file.path('GSE123005_RAW/',i),header = T)
})
lapply(dat, head)
do.call(rbind,lapply(dat, dim))
可以看到,确实是每个txt的行数不一样哦:
> do.call(rbind,lapply(dat, dim))
[,1] [,2]
[1,] 22555 6
[2,] 22555 6
[3,] 22555 6
[4,] 22555 6
[5,] 22548 6
[6,] 22548 6
[7,] 22548 6
[8,] 22548 6
[9,] 22552 6
[10,] 22552 6
[11,] 22552 6
[12,] 22552 6
不过问题不大, 我们取交集即可。每一个txt文件内容节选如下:
gene_short_name refseq_id alternative_refseq_ids locus ko4_FPKM
1 mt-Nd5 unknown none MT:28-16297 1494.49000
2 mt-Tp unknown none MT:28-16297 1.29440
3 Shroom4 NM_001040459 none X:6580843-6583513 1.71424
4 Syp NM_009305 none X:7638703-7653242 1.59568
5 Prickle3 NM_001290624 NM_175097 X:7657362-7671499 7.30217
6 Gpkow NM_173747 none X:7697125-7710323 12.29920
这应该是作者自己流程混乱导致的,很诡异的表达矩阵。
但是我看了看,这个表达矩阵本身就各种冲突:

可以看到,同样的基因,各种ID都是一致的,连坐标都一致,可是居然有两个截然不同的表达量。这个时候并没有最好的部分,我们只能说先往后面走:
dat = lapply(dat, function(x){
# x=dat[[2]]
x=x[x[,5]>0.1,]
x=x[x[,1]!='-',]
x=x[order(x[,1],x[,5],decreasing = T),]
x=x[!duplicated(x[,1]),]
rownames(x)=x[,1]
x
})
ids=unique(unlist( lapply(dat,rownames)))
df = do.call(cbind,
lapply(dat, function(x){
x[ids,5]
}))
head(df)
rownames(df)=ids
colnames(df)=substring(fs,12,14)
head(df)
我看了看,确实这个代码有点复杂,想不出来很正常。
不过呢,最后拿到了一个fpkm矩阵,我感觉也是鸡肋。
这个时候大家需要自己去看文献找到其数据处理方式了,可以看到走的仍然是非常老套的tophat+Cufflinks 流程:
- Cufflinks identified and quantified the transcripts from the preprocessed RNA-Seq alignment- assembly.
- After this, Cuffmerge merged the identified transcript pieces to full length transcripts and annotated the transcripts based on the given annotations.
- Operating on the RNA-Seq alignments and Cufflinks processing, Cuffdiff tracked the mapped reads and determined the fragment per kilobase per million mapped reads (FPKM) for each transcript in all the samples. Primary transcripts and gene FPKMs were then computed by adding up the FPKMs of each primary transcript group or gene group, to determine the differential expression levels at transcript and gene level including a measure of significance between samples / conditions
- Genome_build: mm10 / GRCm38
- Supplementary_files_format_and_content: tab-delimited text files include FPKM values for each Sample
学徒作业
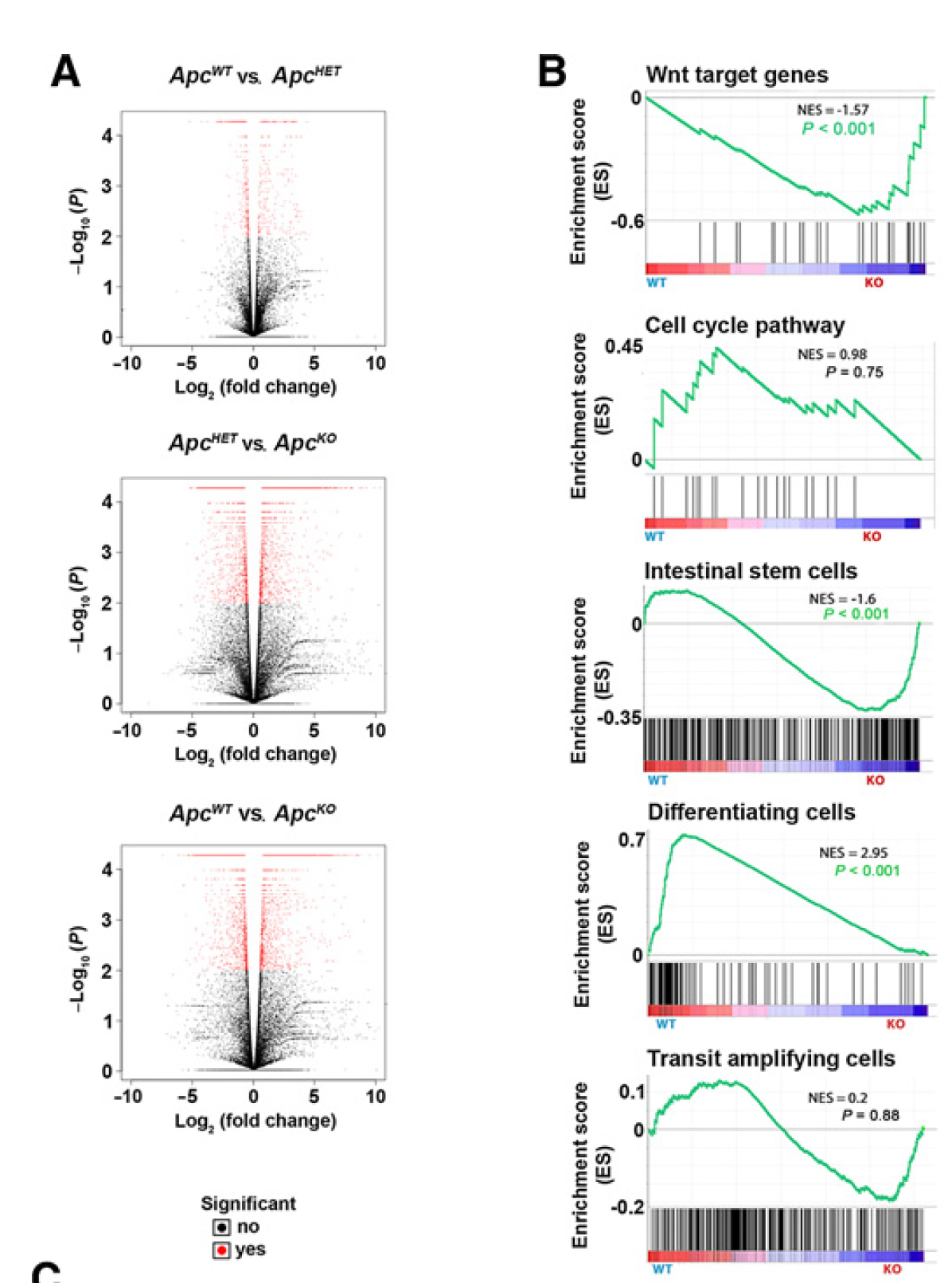
上面的3个分组,两两之间比较,可以做三次差异分析,大家学会了上面的代码后,做出下面的火山图以及指定通路的gsea分析图表:
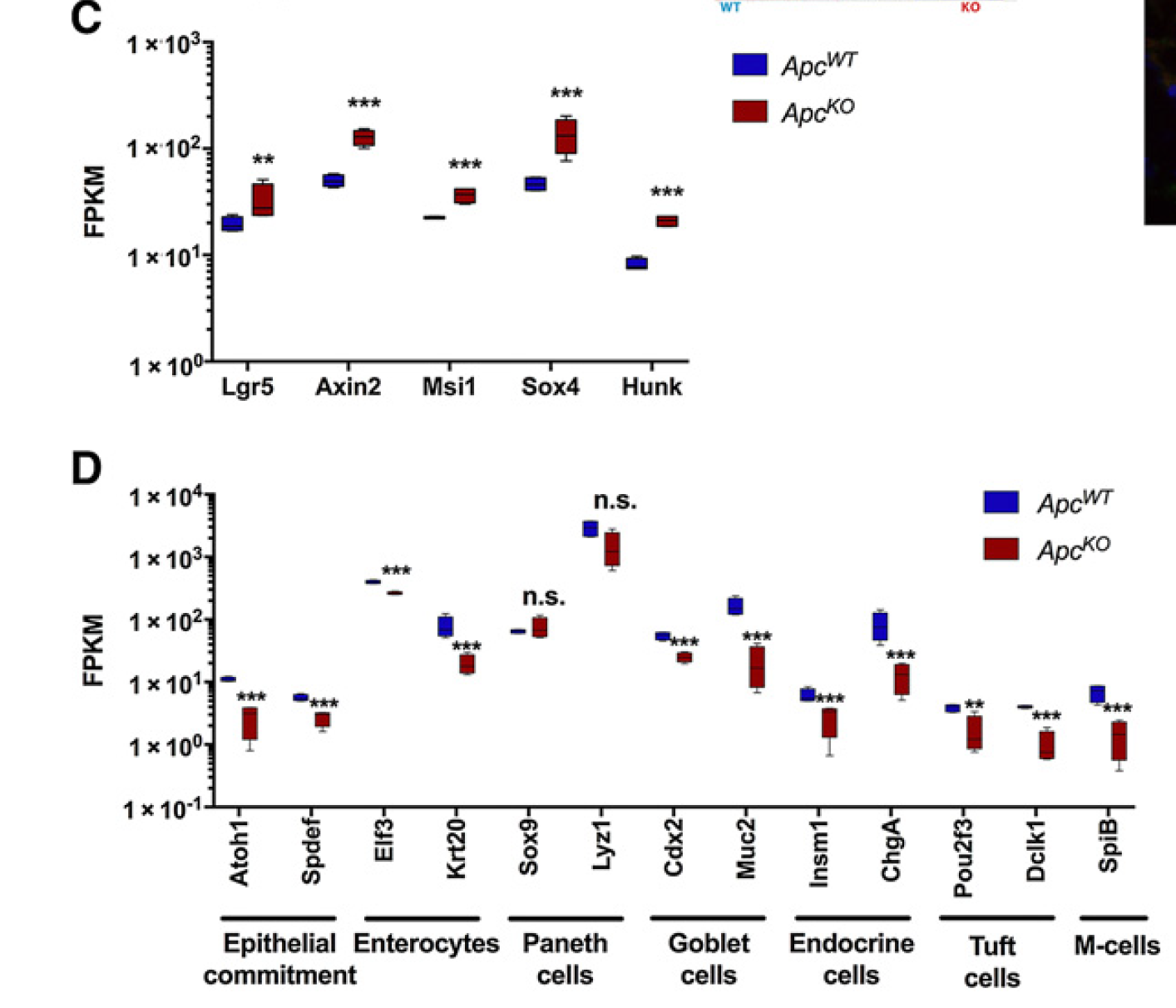
最后,我觉得指定基因的表达量boxplot也很有意思:
一起做一下吧!如果是差异分析,基本上看我五年前的《数据挖掘》系列推文 就足够了;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
更高级作业
拿到 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA507236 的这个项目的原始测序fastq文件,走转录组上游流程,重新定量后拿到counts矩阵哦。
这个需要学习:免费视频课程《RNA-seq数据分析》