我们以 seurat 官方教程为例:
```r
rm(list = ls())
library(Seurat)
devtools::install_github(‘satijalab/seurat-data’)
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
load(file = ‘basic.sce.pbmc.Rdata’)
DimPlot(pbmc, reduction = ‘umap’,
label = TRUE, pt.size = 0.5) + NoLegend()
sce=pbmc
如果你不知道 basic.sce.pbmc.Rdata 这个文件如何得到的,麻烦自己去跑一下 [可视化单细胞亚群的标记基因的5个方法](https://mp.weixin.qq.com/s/enGx9_Sv5wKLdtygL7b4Jw),自己 save(pbmc,file = 'basic.sce.pbmc.Rdata') ,我们后面的教程都是依赖于这个 文件哦!
### 首先对每个细胞亚群找高表达量的标记基因
```r
# 参考: https://mp.weixin.qq.com/s/enGx9_Sv5wKLdtygL7b4Jw
if (file.exists('sce.markers.all_10_celltype.Rdata')) {
load('sce.markers.all_10_celltype.Rdata')
}else {
sce.markers <- FindAllMarkers(object = sce, only.pos = TRUE,
min.pct = 0.25,
thresh.use = 0.25)
save(sce.markers,file = 'sce.markers.all_10_celltype.Rdata')
}
DT::datatable(sce.markers)
library(dplyr)
# 不同seurat版本的 avg_logFC 不一样
top5 <- sce.markers %>% group_by(cluster) %>% top_n(5, avg_logFC)
DoHeatmap(sce,top5$gene,size=3)
普通的热图或者气泡图可视化
DT::datatable(sce.markers)
library(dplyr)
# 不同seurat版本的 avg_logFC 不一样
top5 <- sce.markers %>% group_by(cluster) %>% top_n(5, avg_logFC)
DoHeatmap(sce,top5$gene,size=3)
p <- DotPlot(sce, features = unique(top5$gene) ,
assay='RNA' ) + coord_flip()
p+ theme(axis.text.x = element_text(angle = 45,
vjust = 0.5, hjust=0.5))
如下所示:
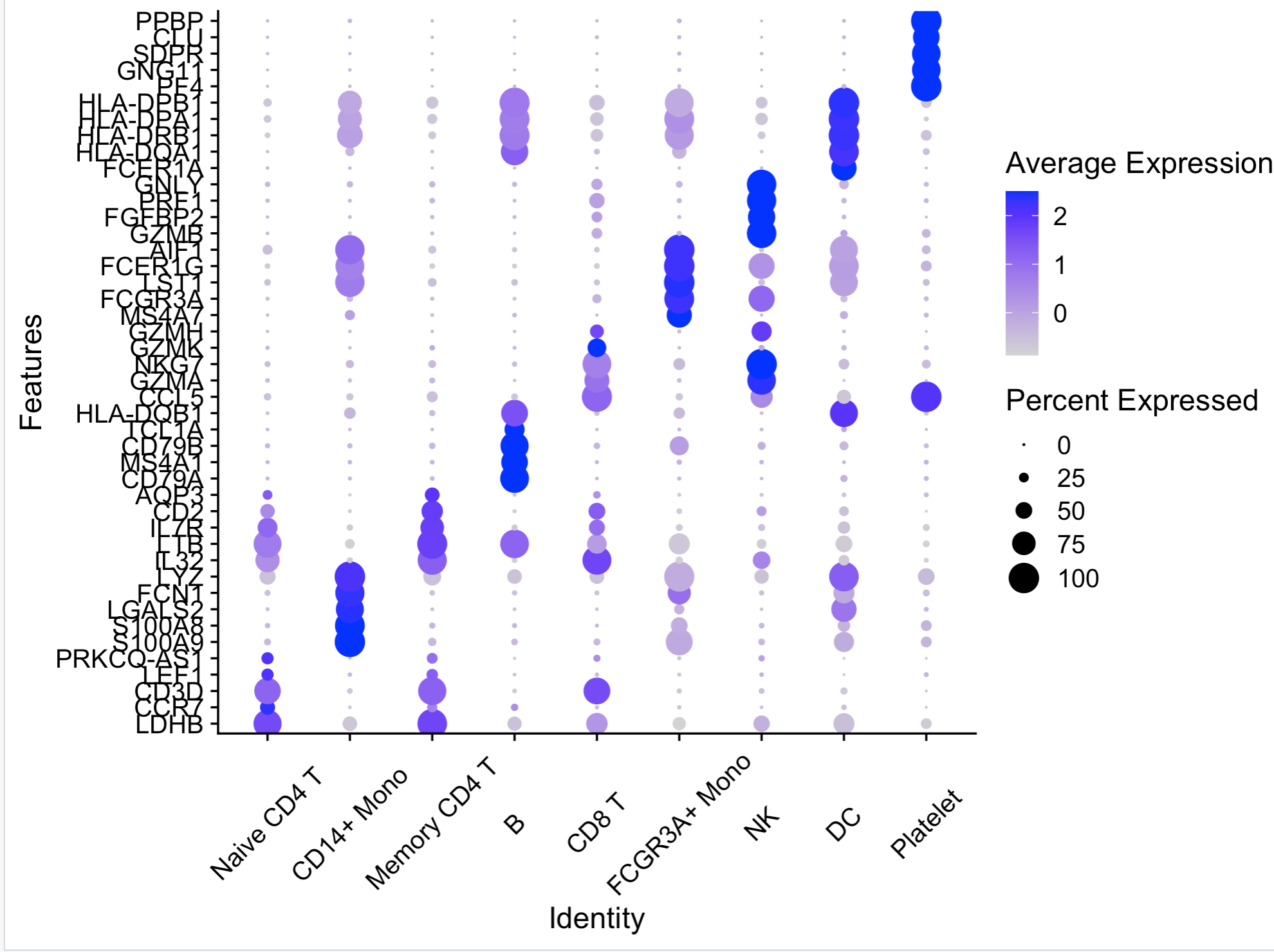
其实这个函数不仅仅是接受一个向量,还可以接受一个列表,代码如下:
head(top5)
top5=top5[!duplicated(top5$gene),]
select_genes_all=split(top5$gene,top5$cluster)
select_genes_all
DotPlot(object = sce,
features=select_genes_all,
assay = "RNA") + theme(axis.text.x = element_text(angle = 45,
vjust = 0.5, hjust=0.5))
出图如下:
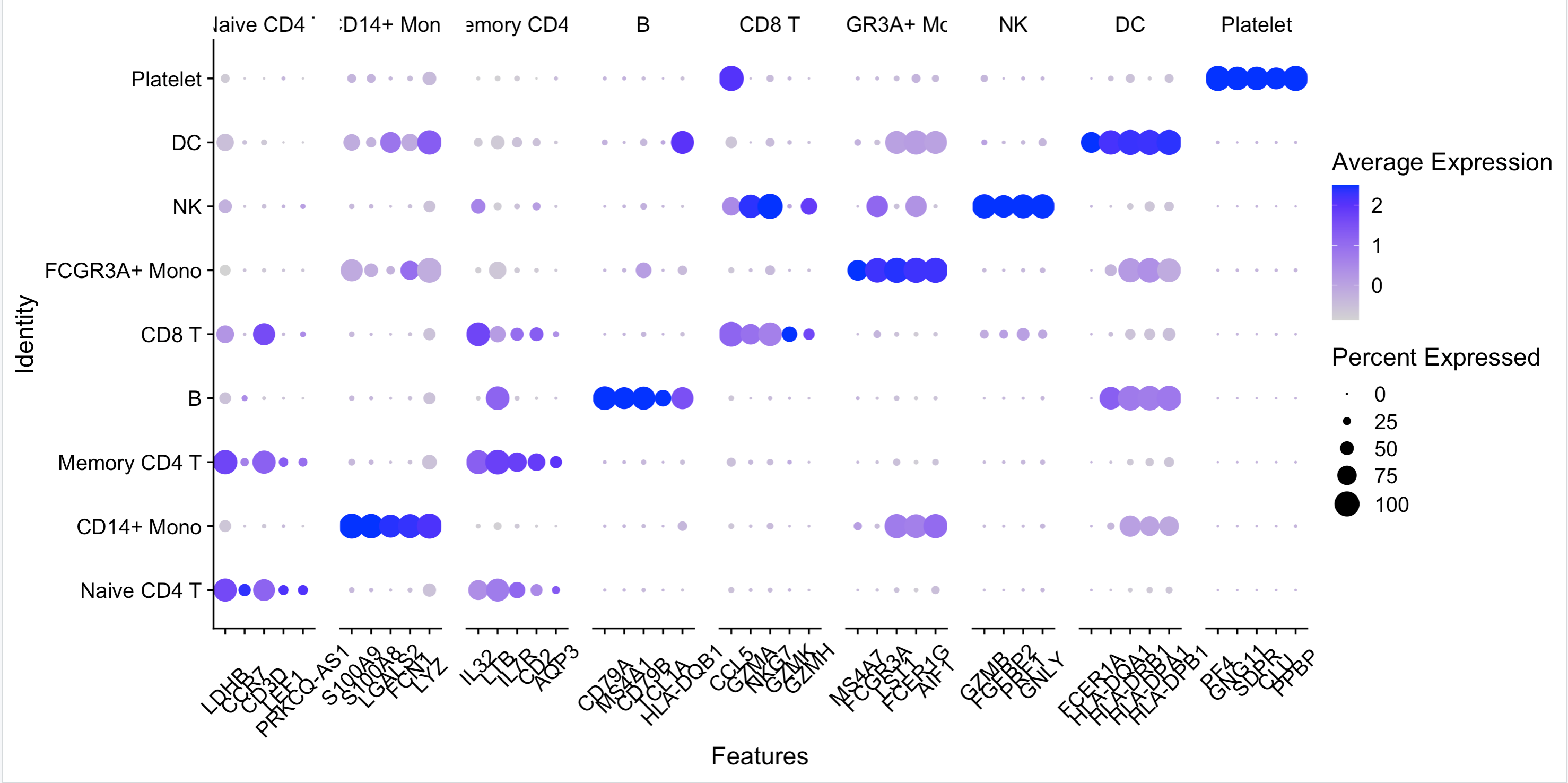
这样的雕虫小技肯定是入不了大家的法眼,出一个作业吧,文章是《High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types》,里面的图 ,见: https://mp.weixin.qq.com/s/-17oUL0-GProZb9apiZJkg