猪马牛羊狗等动物的科学研究貌似并不多,最近接到《生信技能树》公众号后台粉丝提问,跟着我们的转录组课程没办法完成自己的数据分析,因为物种不一样。
我这里以牛来举例吧,说明一下牛的参考转录组序列文件下载及salmon索引构建,以及转录组流程!
首先在 https://m.ensembl.org/Bos_taurus/Info/Annotation 可以查看牛的基因组的基本情况:
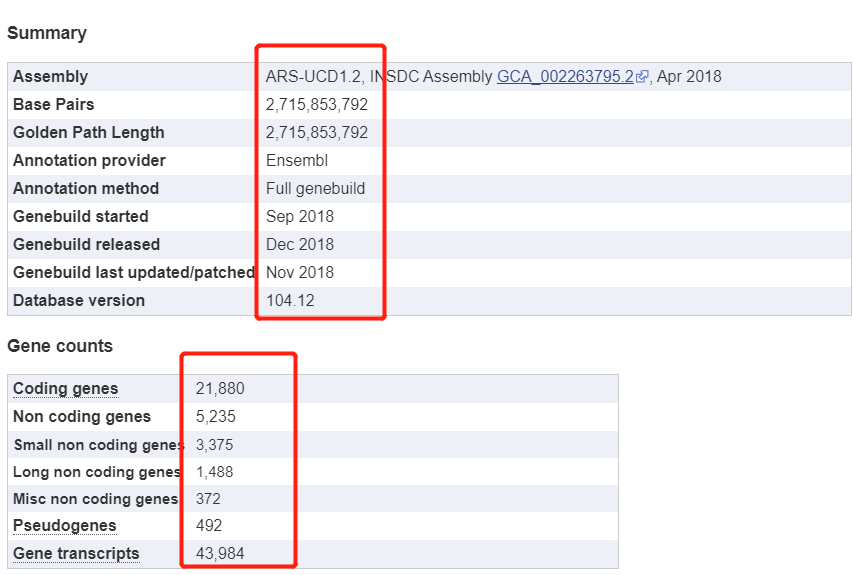
这样就对牛这个物种有一个大概的认知,可以看到牛的参考基因组质量还行,基因注释也是比较不错的!
然后去: http://asia.ensembl.org/Bos_taurus/Info/Index 可以看到参考基因组和参考转录组的下载地址:
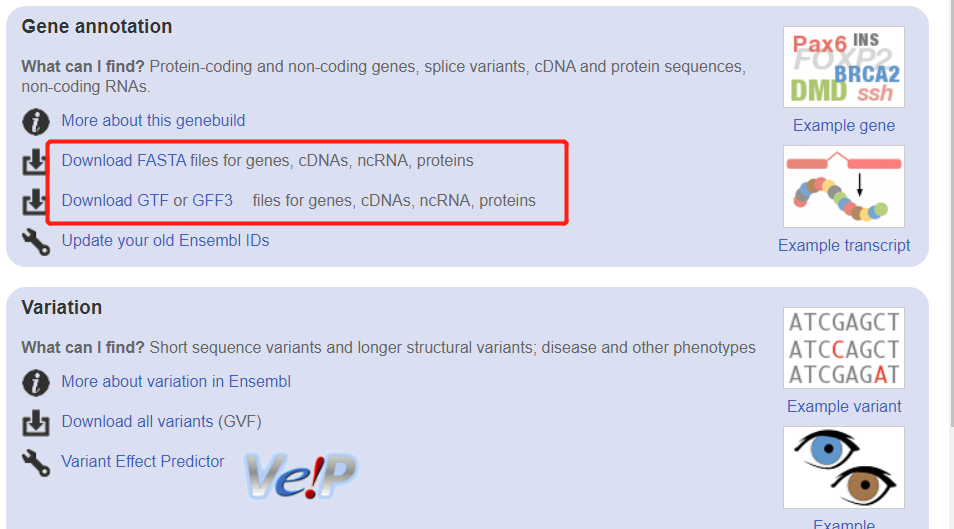
点击进去就可以看到最新版地址了,因为要演示salmon流程,仅仅是需要参考转录组序列的fasta文件即可,代码如下:
wget http://ftp.ensembl.org/pub/release-104/fasta/bos_taurus/cdna/Bos_taurus.ARS-UCD1.2.cdna.all.fa.gz
gunzip Bos_taurus.ARS-UCD1.2.cdna.all.fa.gz
# 100M Mar 28 02:02 Bos_taurus.ARS-UCD1.2.cdna.all.fa
salmon -v
# salmon 0.13.1
# 参考: https://mp.weixin.qq.com/s/kG2uI3me8Xo69mClThRutA
salmon index -t Bos_taurus.ARS-UCD1.2.cdna.all.fa -i Bos_taurus.ARS-UCD1.2.cdna.salmon.index
得到的salmon的index是一个文件夹,内容如下:
ls -lh Bos_taurus.ARS-UCD1.2.cdna.salmon.index/|cut -d" " -f 5-
4.4K May 9 21:17 duplicate_clusters.tsv
793M May 9 21:18 hash.bin
666 May 9 21:18 header.json
115 May 9 21:18 indexing.log
1.5K May 9 21:18 quasi_index.log
77 May 9 21:18 refInfo.json
12M May 9 21:17 rsd.bin
355M May 9 21:17 sa.bin
91M May 9 21:17 txpInfo.bin
96 May 9 21:18 versionInfo.json
单段fastq测序数据的salmon流程
前面的质控就不再赘述,直接上批量salmon流程代码:
## 定量获得TPM值
mkdir salmon_output
cd cleanData
index=../ref/Bos_taurus.ARS-UCD1.2.cdna.salmon.index/
# 单端测序
ls *gz|grep -v "_1" |grep -v "_2" |while read id;do
echo salmon quant -i $index --libType A -r ${id} -o ../salmon_output/${id}_quant --seqBias --gcBias --validateMappings
done > single_salmon.sh
quant.sf文件很重要,要用于后续的分析。ENST和ENSG的前三个字母(ENS),意思是“ENSENMBLE”。T是指转录本。G是指基因。当然,也会看到ENSP,P自然是指蛋白质了。
双端fastq测序数据的salmon流程
index=../ref/Bos_taurus.ARS-UCD1.2.cdna.salmon.index/
# 双端测序
ls *val*gz|cut -d"_" -f 1|sort -u |while read id;do
echo salmon quant -i $index -l ISF --gcBias \
-1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -p 2 \
-o ../salmon_output/${id}_output
done > paired_salmon.sh
这里的 —libType A 参数一定要搞对哦,我就遇到过使用了错误的参数而比对率超级低的情况:
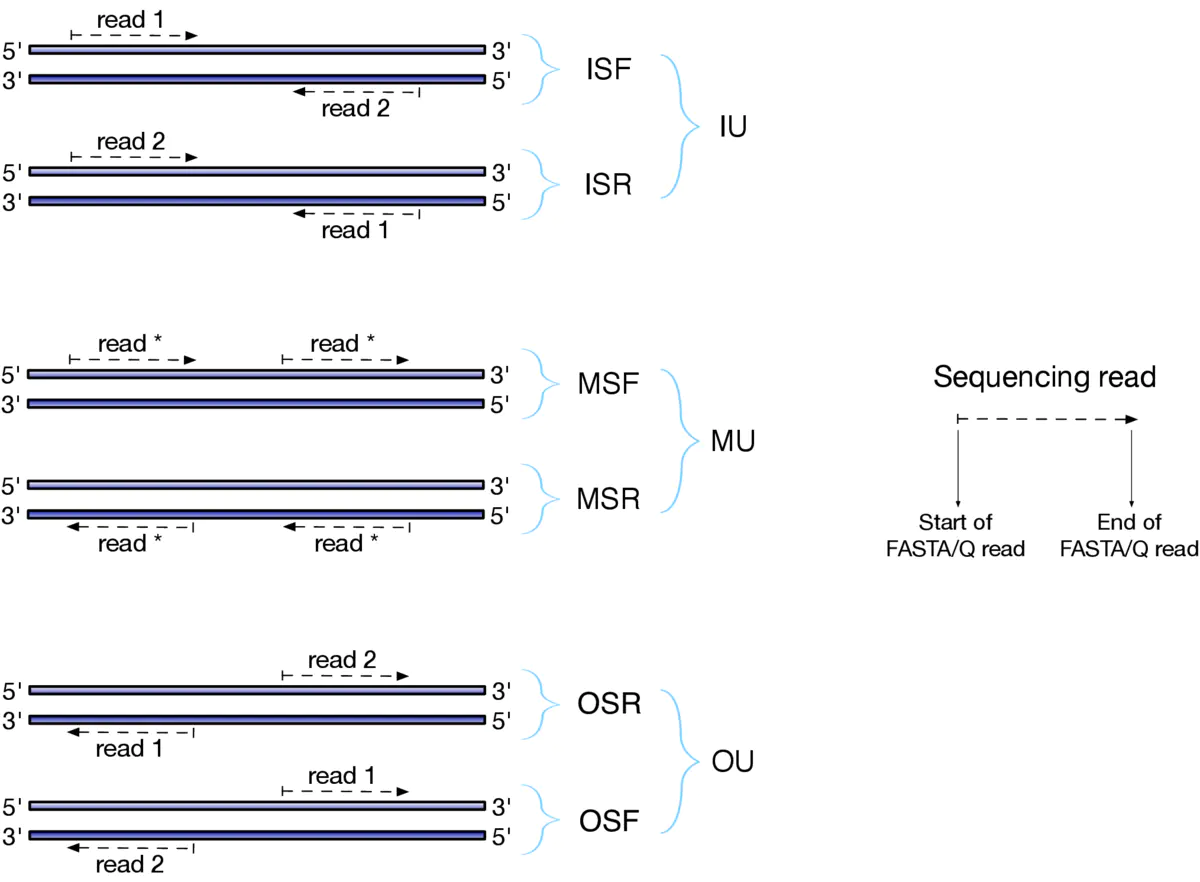
参考教程:http://www.bio-info-trainee.com/2809.html
如果是下载参考基因组和基因注释文件
需要注意的 是 有 3个版本的哦:
<species>: The systematic name of the species.
<assembly>: The assembly build name.
<sequence type>:
* 'dna' - unmasked genomic DNA sequences.
* 'dna_rm' - masked genomic DNA. Interspersed repeats and low
complexity regions are detected with the RepeatMasker tool and masked
by replacing repeats with 'N's.
* 'dna_sm' - soft-masked genomic DNA. All repeats and low complexity regions
have been replaced with lowercased versions of their nucleic base
也多次讲解了,不再赘述,感兴趣的可以去看!基本上就是RNA-seq数据分析路线了,参考我在B站的教学视频,视频观看方式 :
-
视频免费在B站:https://www.bilibili.com/video/BV12s41137HY 大家学习的时候记得发弹幕交流哈。
-
也有微云离线版本视频下载本地播放:
-
- 上游分析视频以及代码资料在:https://share.weiyun.com/5QwKGxi
- 下游主要是基于counts矩阵的标准分析的代码 https://share.weiyun.com/50hfuLi
-
RNA-SEQ实战演练的素材:https://share.weiyun.com/5h1Z2QY ,包括一些公司PPT,综述以及文献以及测试数据
-
RNA-SEQ 实战演练的思维导图:文档链接:https://mubu.com/doc/38y7pmgzLg 密码:p6fo
转录组的标准分析,比较容易复现,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;