最近有粉丝在我们《生信技能树》公众号后台提出来了一个很有意思的问题, 他做的是2X3X3=18个样品的转录组测序,做完了各种各样的组合的差异分析,也做了WGCNA,想多加一个花样,就是最近看到的蛋白编码基因和非编码基因的表达量相关性探索。
因为是转录组测序,所以基因表达量可以很容易根据物种对应的gtf文件进行蛋白编码基因和非编码基因的表达量拆分,前面的代码略过哈,我们直接看拆分好的两个表达量矩阵:
rm(list = ls())
options(stringsAsFactors = F)
load( file = 'input.Rdata')
cg=tail(names(sort(apply(non_matrix, 1, mad))),5000)
non_matrix=non_matrix[cg,]
cg=tail(names(sort(apply(pd_matrix, 1, mad))),5000)
pd_matrix=pd_matrix[cg,]
# 编码基因表达量 是非编码的30倍以上
library(edgeR)
non_matrix=log2(edgeR::cpm(non_matrix)+1)
pd_matrix=log2(edgeR::cpm(pd_matrix)+1)
cor(as.numeric(non_matrix[1,]),
as.numeric(pd_matrix[1,]))
pd_matrix[1:4,1:4]
non_matrix[1:4,1:4]
如下所示:
> pd_matrix[1:4,1:4]
A1 A2 A3 A4
GPALPP1 5.639469 5.529596 5.553193 5.778527
LONP2 7.204279 7.222245 7.201439 7.259283
DYRK1B 4.609923 4.739477 5.049438 4.720654
FBXO44 5.687162 5.277632 5.612554 5.911171
> non_matrix[1:4,1:4]
A1 A2 A3 A4
AL451065.1 3.772673 1.865725 3.484377 4.136150
AL157384.1 3.772673 3.159230 2.830886 3.591639
AL161454.1 2.874547 3.958360 2.830886 2.706604
PTCSC2 1.637491 1.215526 2.343903 2.932325
然后针对拆分好的的蛋白编码基因和非编码基因的表达量矩阵进行相关性计算:
dat=rbind(pd_matrix,non_matrix)
# Unfortunately, the function cor() returns only the correlation coefficients between variables
cor_dat=cor(t(dat))
cor_df=cor_dat[1:nrow(pd_matrix),(nrow(pd_matrix)+1):(nrow(pd_matrix)+nrow(non_matrix))]
round(cor_df[1:4,1:4], 2)
library(reshape2)
df=melt(cor_df)
很有意思的结果,如下所示:
> head(df)
Var1 Var2 value
1 GPALPP1 AL451065.1 0.05102492
2 LONP2 AL451065.1 0.26263048
3 DYRK1B AL451065.1 -0.09633345
4 FBXO44 AL451065.1 0.36313086
5 ZCCHC17 AL451065.1 0.19848955
6 GTF2B AL451065.1 0.15344890
> dim(df)
[1] 25000000 3
> table(abs(df$value) > 0.9)
FALSE TRUE
24845266 154734
> table(abs(df$value) > 0.99)
FALSE TRUE
24999890 110
也就是说我们的五千个蛋白编码基因和五千个非编码基因两两组合是可以有250万个相关性,如果使用0.9的相关性作为阈值来挑选那些蛋白编码基因与非编码基因表达量正相关或者负相关的组合,会出15万个!当然了,如果是使用0.99这样的严苛阈值,可以筛选到只有110个相关性基因对。但是,我检查了其中一个,发现非常的有趣 :
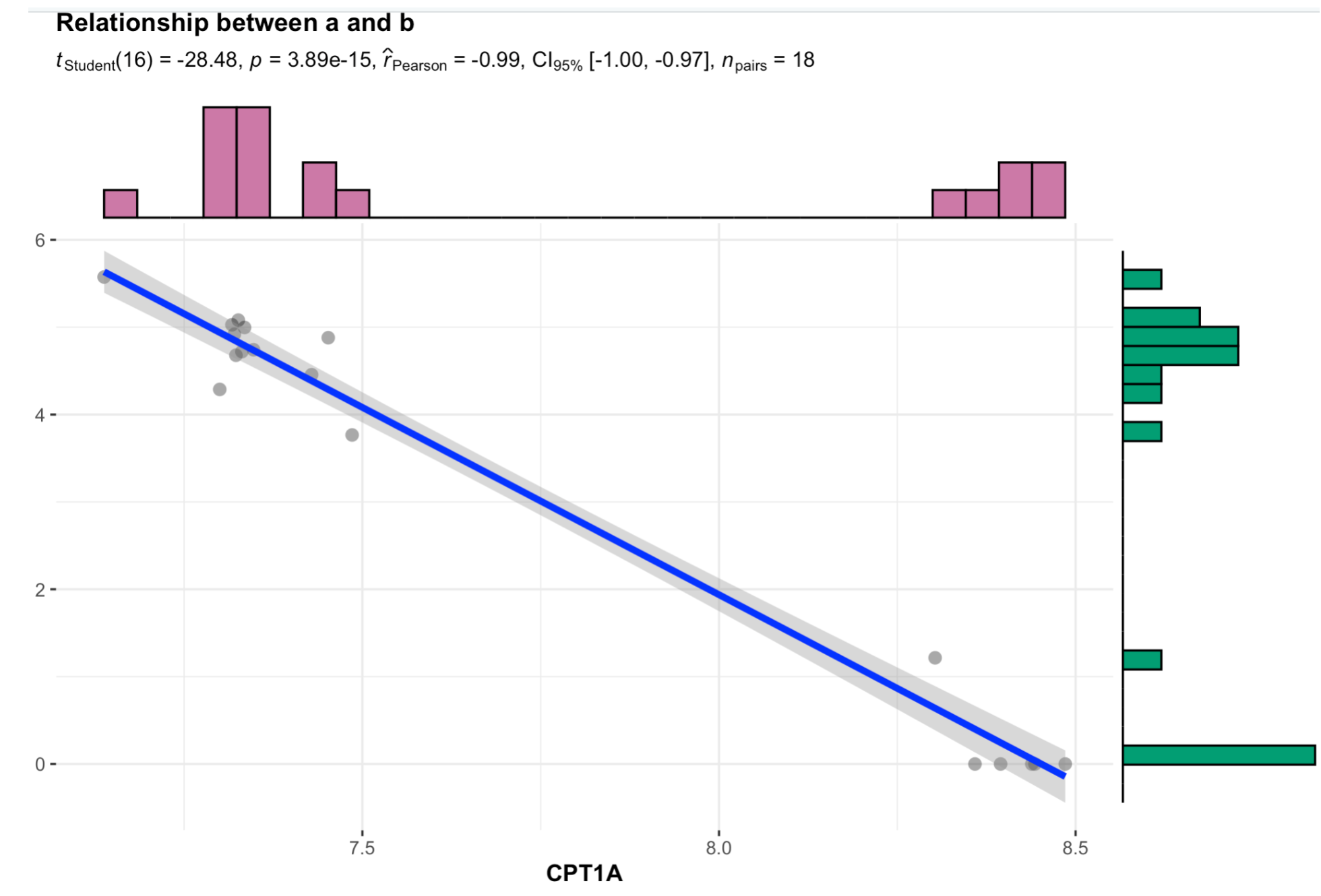
如果你用肉眼看,其实这两个基因的表达量并没有太好的相关性,它们之所以被计算公式得到了这么高的相关性,仅仅是因为我们的18个样品可以分成泾渭分明的两个组,只要是在这两个组有表达量差异的基因,就容易被算出来有表达量相关性!
绘图代码如下:
library(ggstatsplot)
colnames(dat)
dat=as.data.frame(t(dat))
colnames(dat)
ggscatterstats(data = dat ,
y = AC079949.5,
x = CPT1A,
centrality.para = "mean",
margins = "both",
xfill = "#CC79A7",
yfill = "#009E73",
marginal.type = "histogram",
title = "Relationship between a and b")
如果我把这两个基因的表达量的相关性根据在18个样品里面的表达量拆分成为高低两个分组后再计算相关性:
x=dat[,'AC079949.5']
y=dat[,'CPT1A']
plot(y,x)
kp=x>2
df=data.frame(x,y,kp)
head(df)
library(ggpubr)
ggscatter(df, x = "y", y = "x",
add = "reg.line", # Add regression line
conf.int = TRUE, # Add confidence interval
color = "kp", palette = "jco", # Color by groups
shape = "kp" # Change point shape by groups
)+ stat_cor(aes(color = kp), label.x = 8) # Add correlation coefficient
可以看到这样的相关性,就没有前面的0.99那样的超级相关性了。
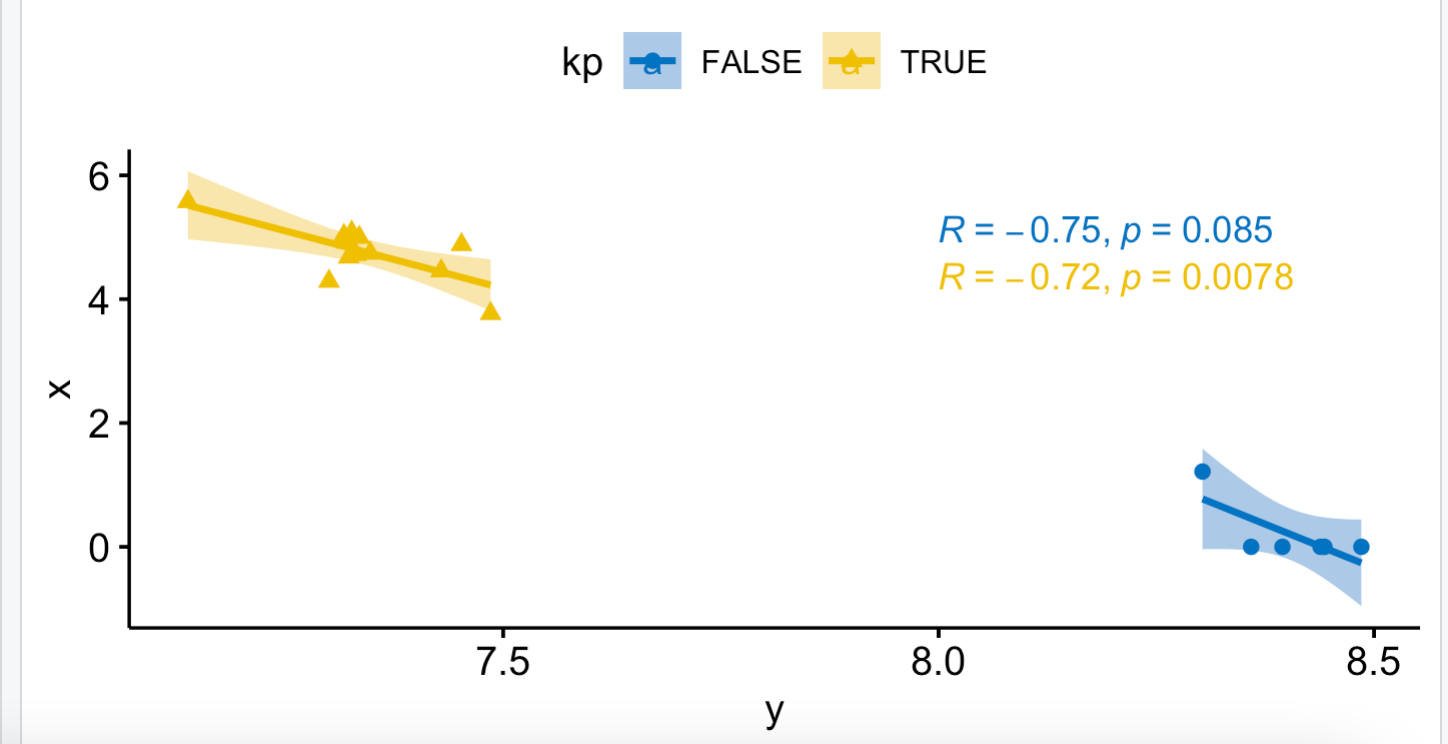
这样就很尴尬了,本来18个样品就分成2X3的6个组,不同处理不同细胞系有差异很正常, 但是这样再计算相关性,其实就很麻烦了。如果你统计学基础知识不够,就会被表面结果给蒙骗!
推荐《故事中的统计学》
这个实战案例,我是在小伊老师的《故事中的统计学》看到的,早期我在进行《生信五周年》巡讲的时候,多次提到了:
不过,能亲耳听到小伊老师的《故事中的统计学》课程的并不多,所以我以《生信五周年》名义邀请小伊老师以视频直播互动形式分享其中的5到10个好玩的,而且能跟咱们生物信息学紧密结合的统计学故事!
进群方式
如果你也感兴趣,欢迎加入哈!老规矩,群聊组建费用18.8元,一个简单的门槛隔绝那些不怀好意的广告营销号!
前200名可以直接扫描群聊二维码进群,满200人后我们会统一收款!(18.8 元即可,如果你不同意这个象征性收费,请不要进群哈!)

如果上面的二维码无法进群,说明已经满员了,需要我们生信技能树的官方拉群小助手帮忙拉群哦!!!(名额有限,先到先得!!!)
这个时候请直接付款28元给小助手,就可以进群,或者你转发此推文到朋友圈然后截图给小助手,就可以仍然以18.8元进群!