前面的教程:[大样本量多分组表达量矩阵分析你难道没想到单细胞吗](https://mp.weixin.qq.com/s/p2oYAgG-LO9yLGx1r3i9zQ),提到了我们整合全部的33种癌症的仅仅是蛋白质编码基因的表达量矩阵,进行降维聚类分群可以看到并不是严格的各个癌症泾渭分明。而去还别出心裁的引入了单细胞经典seurat流程,进行降维聚类分群。
这个时候眼尖的小伙伴就指出来了,我们并没有区分表达量矩阵里面的正常组织和肿瘤组织,哪怕是不同的癌症。其实在tcga数据挖掘时候很容易区分, 我们主要是使用如下所示代码:
```r
library(Seurat)
load(file = 'sce.Rdata')
gp=substring(colnames(sce),14,15)
table(gp)
sce@meta.data$gp=gp
library(gplots)
balloonplot(table(sce@meta.data$gp,sce@meta.data$group))
```
首先看看是不是所有的癌症都有正常对照:

然后汇总一下,可以看到:
```
> as.data.frame(table(gp))
gp Freq
1 01 9724
2 02 47
3 03 151
4 05 11
5 06 393
6 07 1
7 11 730
```
大家可以去:https://docs.gdc.cancer.gov/Encyclopedia/pages/TCGA_Barcode/ 查看这个 TCGA数据样本编码Barcode的意义,由横杠连接的7个子编码信息组成,代表了详细的临床样本收集,检测,分析信息,包括项目名称(**Project**),研究机构(**TSS**),受检者编号(**Participant**),样本组织类型(**Sample**)(常用的是01代表实体瘤和11代表实体瘤旁),样本顺序编号**(Vial**),样本顺序子编号(**Portion**),分析的分子类型编码比如DNA,RNA(**Analyte**),样本在96孔板中的顺序编号(**Plate**),检测的数据分析机构(**Center**)。
我们这个时候选取了第14和15位数字, 就是样本组织类型(**Sample**)(常用的是01代表实体瘤和11代表实体瘤旁) ,详见: https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/sample-type-codes
接下来就可以进行简单可视化:
```r
library(patchwork)
p1=DimPlot(sce,reduction = "tsne",label=T,repel = T,
group.by ='gp')
p2=DimPlot(sce,reduction = "umap",label=T,repel = T,
group.by ='gp')
p1+p2
```
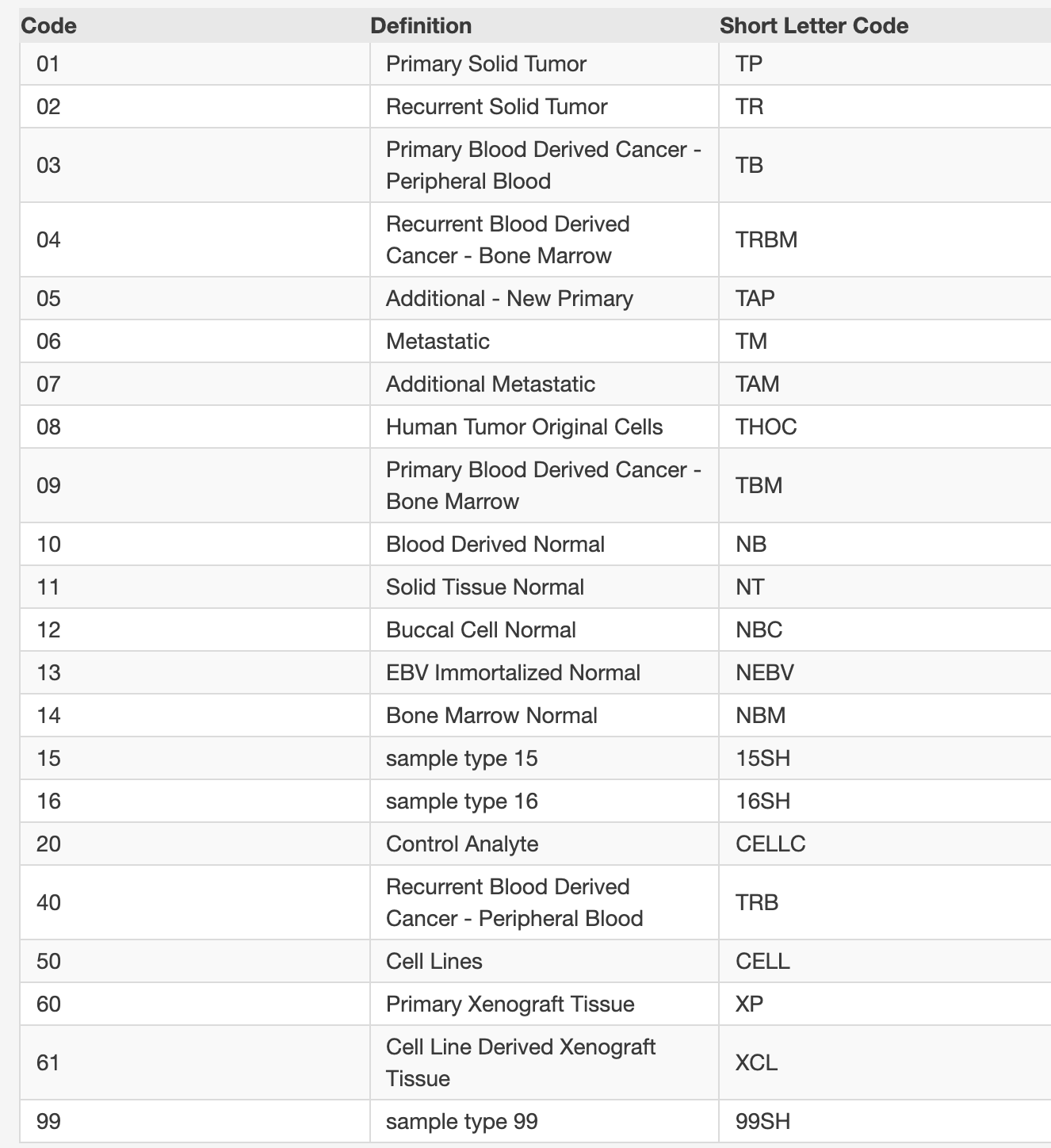
可以看到,正常对照在每个癌症内部:
但是这个默认配色有点尴尬,这里顺便介绍一个 神器啊,https://colorbrewer2.org/ ,我们这里是7个分组,所以自己挑选一个配色就可以输出7个颜色啦!
加上自定义配色后,重新出图:
```r
cl=c('#7fc97f','#beaed4','#fdc086','#ffff99','#386cb0','#f0027f','#bf5b17')
p1=DimPlot(sce,reduction = "tsne",label=T,repel = T,
cols = cl,
group.by ='gp')
p2=DimPlot(sce,reduction = "umap",label=T,repel = T,
cols = cl,
group.by ='gp')
p1+p2
```
效果如下所示:
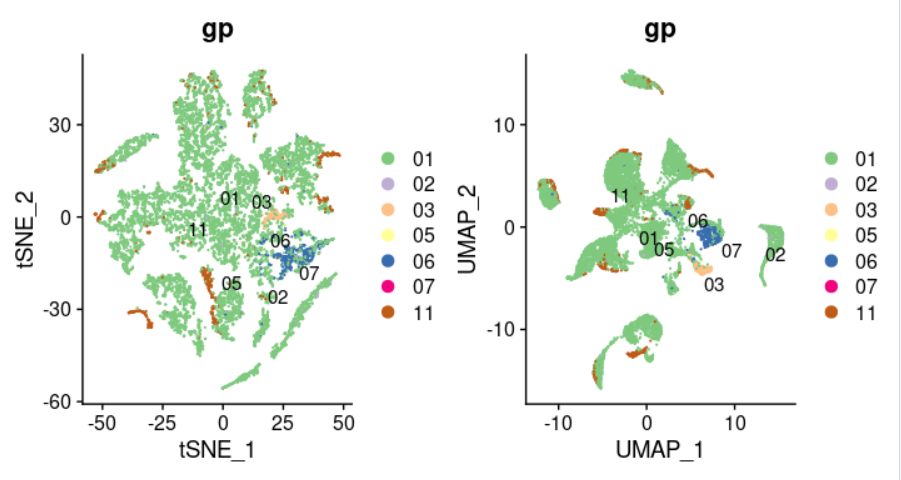
这个时候,11这个数字所代表的就是癌旁这样的正常组织转录组数据,它基本上散步在各个癌症内部,并没有独立成为一个亚群!
如果是跟seurat的标准降维聚类分群进行对比,也可以看到:
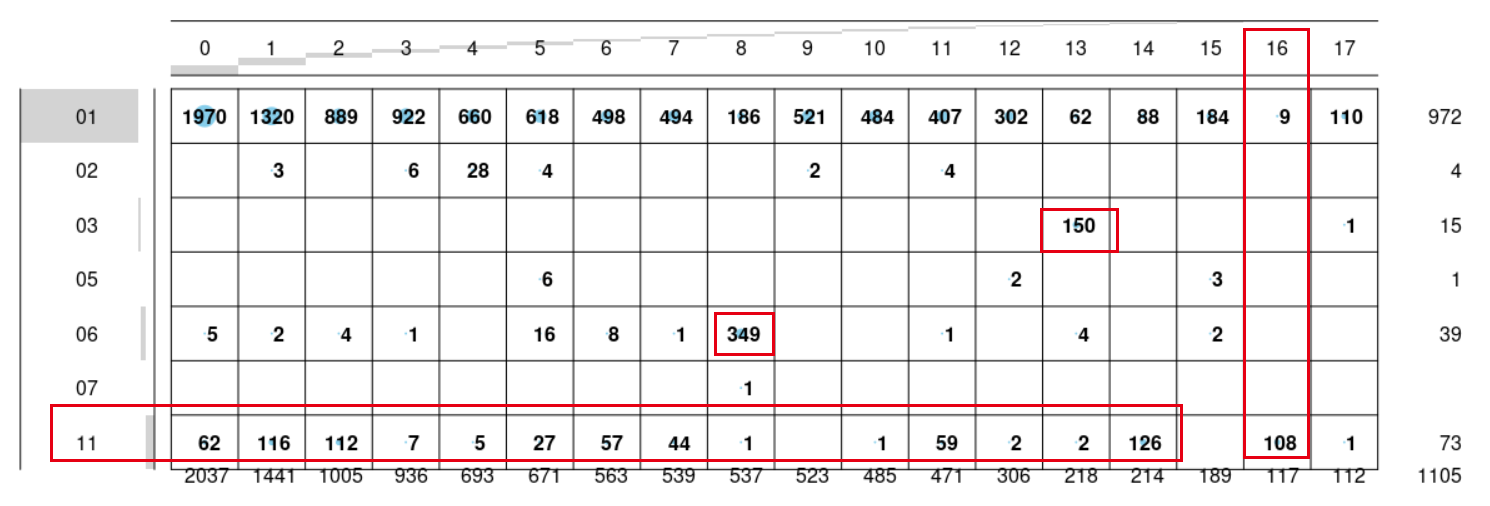
只有这个16群是蛮纯粹的癌旁亚群,我们仔细看看它具体是什么癌症!
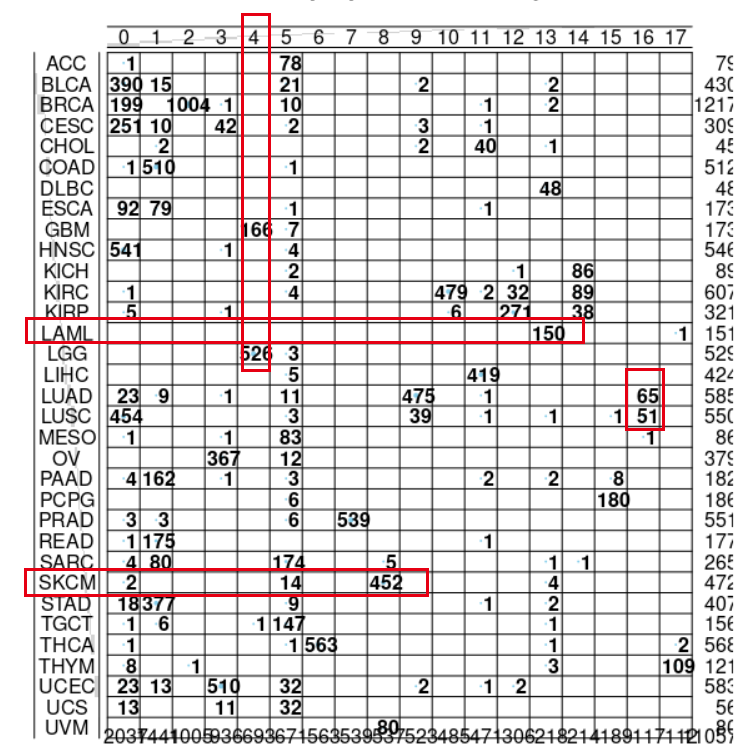
挺有意思的:
- 第16群,是肺癌的正常对照,包括肺鳞癌和非腺癌,是因为肺部特异性很大?只有肺癌的正常组织独立成群,其它癌症的正常组织都紧密结合在各自的癌症部位。
- 第13群都是03,代表Primary Blood Derived Cancer - Peripheral Blood,有因为它不是实体肿瘤,是血液瘤,所以对应LAML 这个癌症
- 第4群,是GBM和LGG,脑癌,它特异性的对应了02个标签,代表Recurrent Solid Tumor
- 而第8群,是SKCM,它里面的06标签最多,是Metastatic,也就是说皮肤癌取样的时候容易采集到转移瘤部位?