经过了大量的单细胞转录组数据分析基础讲解,相信大家对第一层次降维聚类分群都不陌生了。参考我们的《明码标价》专栏里面的单细胞内容
如下所示的一个数据集处理,我们会得到如下所示:
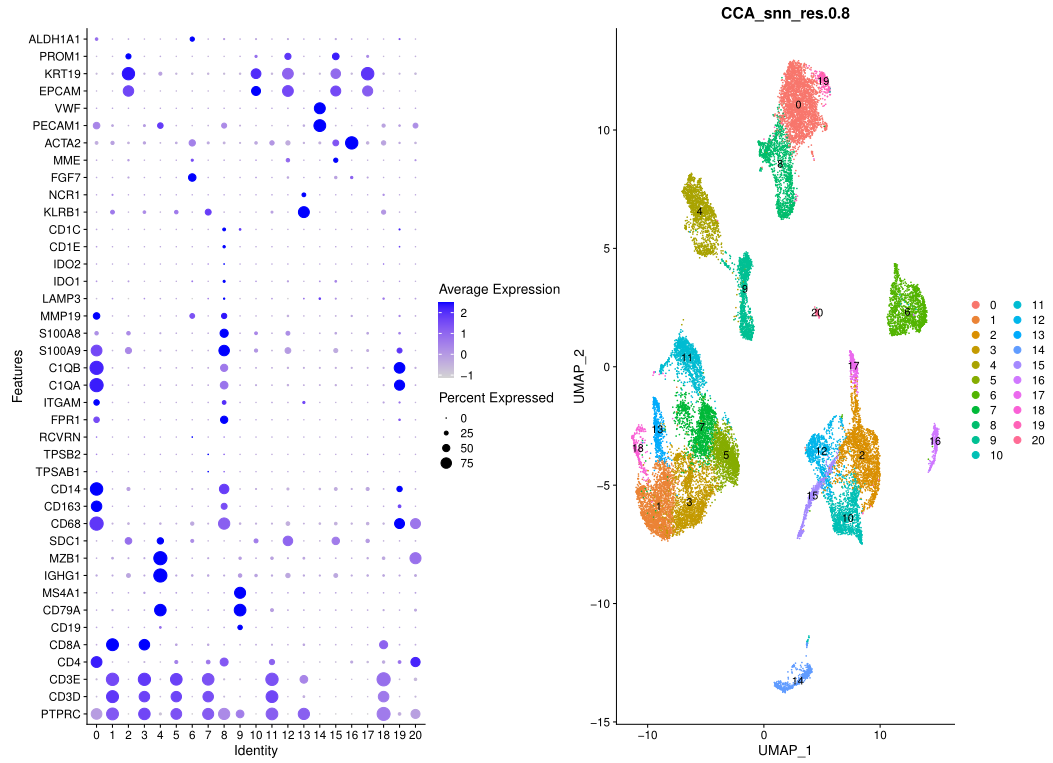
特定的基因名字对应的特定的生物学亚群,如下所示:
'CD68', 'CD163', 'CD14', # myeloids
'TPSAB1' , 'TPSB2', # mast cells,
'RCVRN','FPR1' , 'ITGAM' ,
'C1QA', 'C1QB', # mac
'S100A9', 'S100A8', 'MMP19',# monocyte
'LAMP3', 'IDO1','IDO2',## DC3
'CD1E','CD1C', # DC2
'KLRB1','NCR1', # NK
'FGF7','MME', 'ACTA2', ## fibo
'DCN', 'LUM', 'GSN' , ## mouse PDAC fibo
'Amy1' , 'Amy2a2', # Acinar_cells
'PECAM1', 'VWF', ## endo
'EPCAM' , 'KRT19', 'PROM1', 'ALDH1A1' # epi
简单的肉眼就可以粗略进行各自细胞亚群合并后的生物学命名,如下所示:
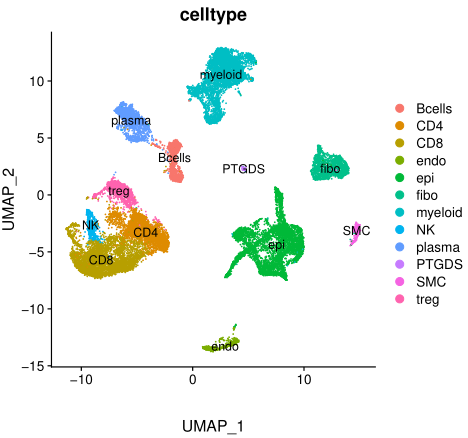
但是这样的分析太常规了,远不够!一般来说, 需要进行各自细胞亚群独立进行更加细致的降维聚类分群:
#拆分为 多个 seurat子对象
sce.all.list <- SplitObject(sce.all , split.by = "celltype")
sce.all.list
names(sce.all.list)
for (i in names(sce.all.list)) {
epi_mat = sce.all.list[[i]]@assays$RNA@counts
epi_phe = sce.all.list[[i]]@meta.data
sce=CreateSeuratObject(counts = epi_mat,
meta.data = epi_phe )
sce
table(sce@meta.data$orig.ident)
save(sce,file = paste0(i,'.Rdata'))
}
得到如下所示各个亚群的rdata文件,都存放在 Rdata-celltype/ 文件夹里面 :
$ ls -lh Rdata-celltype/|cut -d" " -f 5-
5.6M Aug 22 09:39 Bcells.Rdata
14M Aug 22 09:39 CD4.Rdata
22M Aug 22 09:39 CD8.Rdata
3.2M Aug 22 09:40 NK.Rdata
1.3M Aug 22 09:39 PTGDS.Rdata
2.4M Aug 22 09:40 SMC.Rdata
26K Aug 22 09:39 check_stromal_marker_by_RNA_snn_res.0.8.pdf
5.8M Aug 22 09:40 endo.Rdata
63M Aug 22 09:39 epi.Rdata
13M Aug 22 09:39 fibo.Rdata
41M Aug 22 09:40 myeloid.Rdata
6.8M Aug 22 09:39 plasma.Rdata
7.2M Aug 22 09:39 treg.Rdata
继续降维聚类分群
这里我们拿fibo亚群举例:
rm(list=ls())
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(dplyr)
load('../Rdata-celltype/fibo.Rdata')
table(sce@meta.data$orig.ident)
sce
library(stringr)
sce <- NormalizeData(sce, normalization.method = "LogNormalize",
scale.factor = 1e4)
GetAssay(sce,assay = "RNA")
sce <- FindVariableFeatures(sce,
selection.method = "vst", nfeatures = 2000)
sce <- ScaleData(sce)
sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
DimHeatmap(sce, dims = 1:12, cells = 100, balanced = TRUE)
ElbowPlot(sce)
sce <- FindNeighbors(sce, dims = 1:15)
sce <- FindClusters(sce, resolution = 0.8)
table(sce@meta.data$RNA_snn_res.0.8)
set.seed(123)
sce <- RunTSNE(object = sce, dims = 1:15, do.fast = TRUE)
DimPlot(sce,reduction = "tsne",label=T)
sce <- RunUMAP(object = sce, dims = 1:15, do.fast = TRUE)
DimPlot(sce,reduction = "umap",label=T)
head(sce@meta.data)
DimPlot(sce,reduction = "umap",label=T,group.by = 'orig.ident')
sce <- FindClusters(sce, resolution = 0.1)
DimPlot(sce,reduction = "umap",label=T)
ggsave(filename = 'umap_recluster_by_0.1.pdf')
DimPlot(sce,reduction = "umap",label=T,
group.by = 'orig.ident')
ggsave(filename = 'umap_sce_recluster_by_patients.pdf')
table(sce@meta.data$orig.ident,sce@meta.data$seurat_clusters)
colnames(sce@meta.data)
可以看到,如下所示的非常尴尬的降维聚类分群结果:
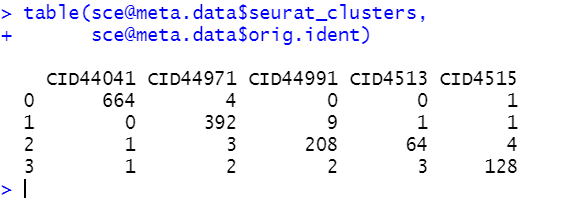
也就是说这个时候区分出来的每个亚群都是各自的病人特异的,有点类似于肿瘤细胞的特性。
如下所示:
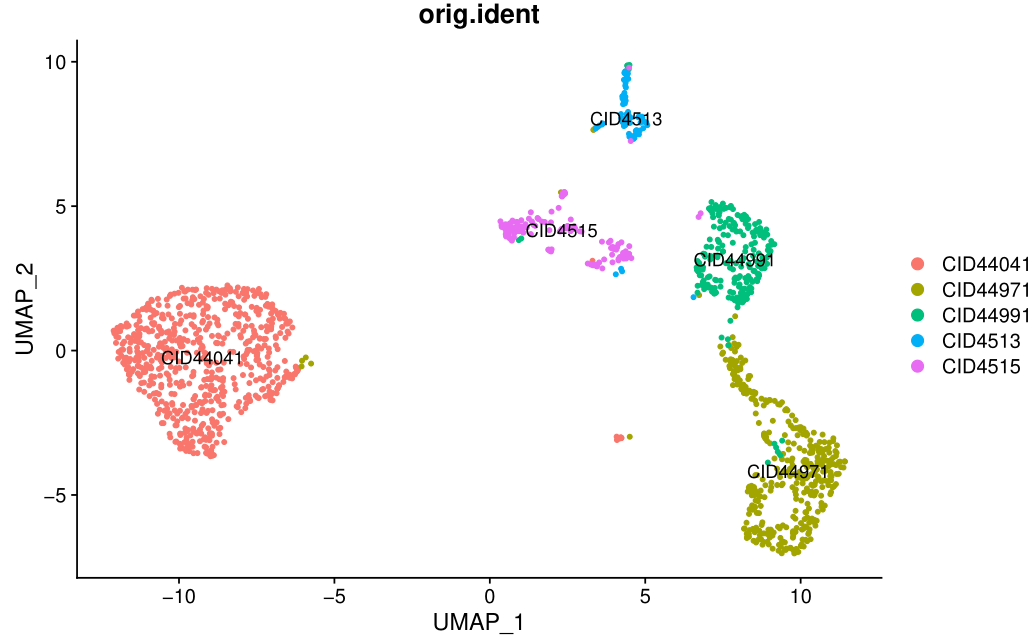
而且常见fibo相关的基因都不特征性表达:
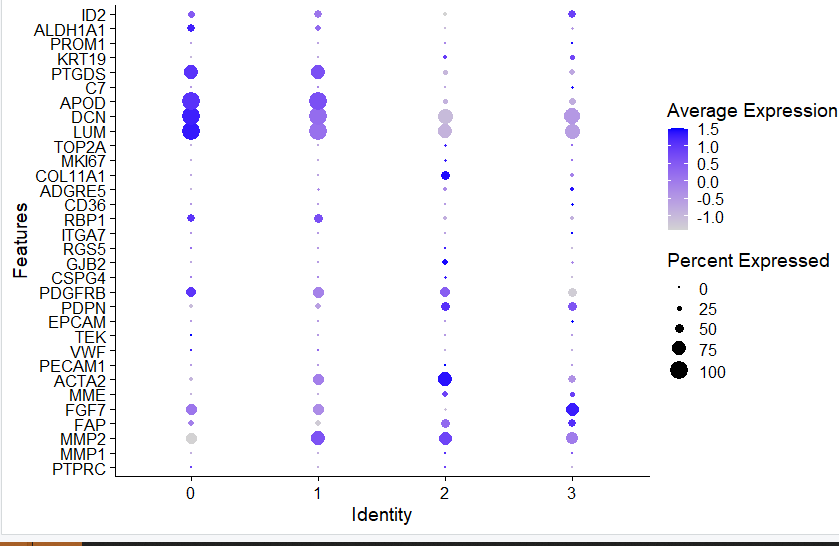
这个时候根本就没办法对这些细胞亚群进行生物学命名。
强烈建议这个时候使用harmony进行整合
代码如下所示;
library(harmony)
seuratObj <- RunHarmony(sce, "orig.ident")
names(seuratObj@reductions)
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
DimPlot(seuratObj,reduction = "umap",label=T )
p1.compare=plot_grid(ncol = 3,
DimPlot(sce, reduction = "pca", group.by = "orig.ident")+NoAxes()+ggtitle("PCA"),
DimPlot(sce, reduction = "tsne", group.by = "orig.ident")+NoAxes()+ggtitle("tSNE"),
DimPlot(sce, reduction = "umap", group.by = "orig.ident")+NoAxes()+ggtitle("UMAP"),
DimPlot(seuratObj, reduction = "pca", group.by = "orig.ident")+NoAxes()+ggtitle("PCA"),
DimPlot(seuratObj, reduction = "tsne", group.by = "orig.ident")+NoAxes()+ggtitle("tSNE"),
DimPlot(seuratObj, reduction = "umap", group.by = "orig.ident")+NoAxes()+ggtitle("UMAP")
)
p1.compare
ggsave(plot=p1.compare,filename="before_and_after_inter.pdf")
sce=seuratObj
sce <- FindNeighbors(sce, reduction = "harmony",dims = 1:15)
sce <- FindClusters(sce, resolution = 0.1)
table(sce@meta.data$seurat_clusters)
DimPlot(sce,reduction = "umap",label=T)
ggsave(filename = 'harmony_umap_recluster_by_0.1.pdf')
DimPlot(sce,reduction = "umap",label=T,
group.by = 'orig.ident')
ggsave(filename = 'harmony_umap_sce_recluster_by_patients.pdf')
整合前后效果很明显:
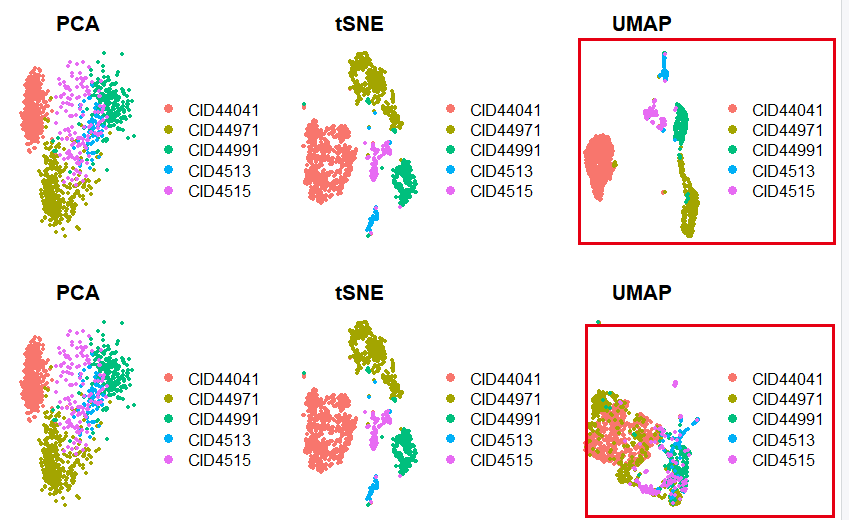
接下来再看不同细胞亚群,就不再倾向于属于某个样品了:
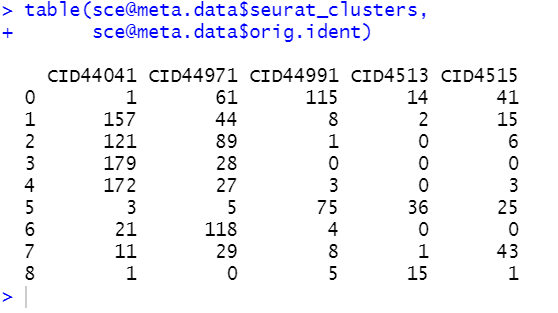
这样的结果更合理,我们的fibo亚群,就可以细分成为不同的而且是跨越了多个病人的亚群,可以对每个亚群找各自高表达基因,并且进行生物学命名。
单细胞转录组数据分析的标准降维聚类分群,并且进行生物学注释后的结果。可以参考前面的例子:人人都能学会的单细胞聚类分群注释 ,我们演示了第一层次的分群。
如果你对单细胞数据分析还没有基础认知,可以看基础10讲: