我看到了一个研究可能对大家有帮助,《Transcriptional response profiles of paired tumor-normal samples offer novel perspectives in pan-cancer analysis》,
发表这个研究的杂志不怎么样, Oncotarget. 2017; 8:41334-41347. https://doi.org/10.18632/oncotarget.17295 但是里面有一个信息蛮有意思的,而且毕竟是比较早期的数据挖掘了,不同于现在的粗制滥造各种灌水套路,起码他们是实打实的做了不少原创性工作了的!
如下所示,这个数据挖掘研究纳入了TCGA数据库里面的样本量还算比较多的癌症种类,而且挑选那些提供了配对样品的 :
- bladder urothelial carcinoma (BLCA, n = 19),
- breast cancer (BRCA, n = 111),
- colon adenocarcinoma (COAD, n = 41),
- head and neck squamous cell carcinoma (HNSC, n = 41),
- kidney chromophobe renal cell carcinoma (KICH, n = 25),
- kidney clear cell renal cell carcinoma (KIRC, n = 72),
- kidney renal papillary cell carcinoma (KIRP, n = 32),
- liver hepatocellular carcinoma (LIHC, n = 50),
- lung adenocarcinoma (LUAD, n = 57),
- lung squamous cell carcinoma (LUSC, n = 51),
- prostate adenocarcinoma (PRAD, n = 52),
- thyroid carcinoma (THCA, n = 59),
- and uterine corpus endometrial carcinoma (UCEC, n = 23)
可以看到,绝大部分癌症的配对样品都不多哦!
亚型之间差异分析然后聚类
首先对每个癌症的这些样品做 paired tumor and normal samples. 的 差异分析,然后挑选 Genes with log2(fold-change) ≥ 2 in at least 10% of all samples were retained for subsequent analysis. 差异分析相信大家都不陌生了,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
经过一系列的差异分析后,挑选合适的基因,以较小的表达量矩阵进入后续的Consensus clustering result at k = 10 分析,如下所示的降维聚类分群结果,是不是跟单细胞数据分析有点类似?
首先是挑选基因,然后是降维聚类分群,然后看不同群里面的不同癌症领域是否聚在一起或者泾渭分明!
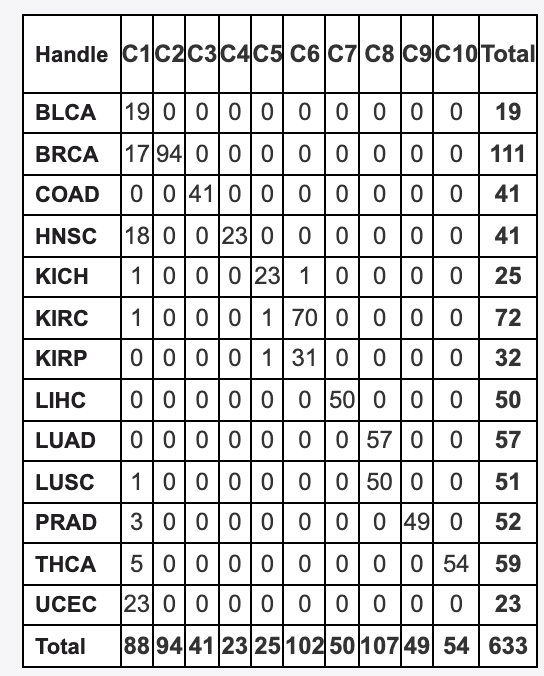
可以看到HNSC被拆分到了C1和C4两个独立的亚群,这个也是作者最重要的结论!其实没有太大的意思,头颈癌里面本来就是不同癌症的混合体,包括口腔癌,鼻咽癌,喉癌等等。
亚群之间差异分析
这个时候的差异分析,不再是各个肿瘤内部的N-T配对差异分析啦,是针对10个亚群,每个都是继续看其相当于其它9个亚群的 差异上下调基因,同样的阈值筛选。如下所示交集:
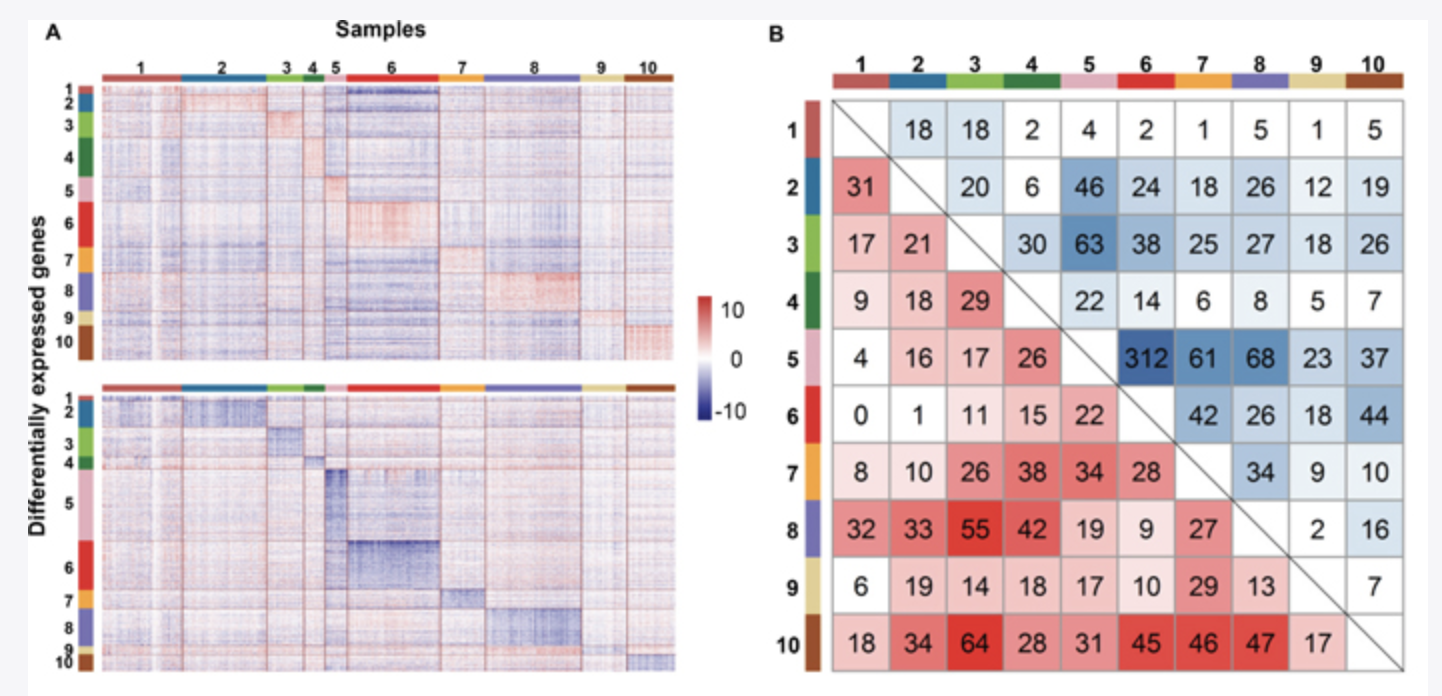
这个步骤,在单细胞数据分析里面超级常见,其实就是每个亚群找标记基因的策略。
针对HNSC
前面提到了HNSC被拆分到了C1和C4两个独立的亚群:
HNSC Subtype 1 (17 samples in C1) and HNSC Subtype 2 (23 samples in C4).
所以就可以进行细分亚群的差异分析啦!
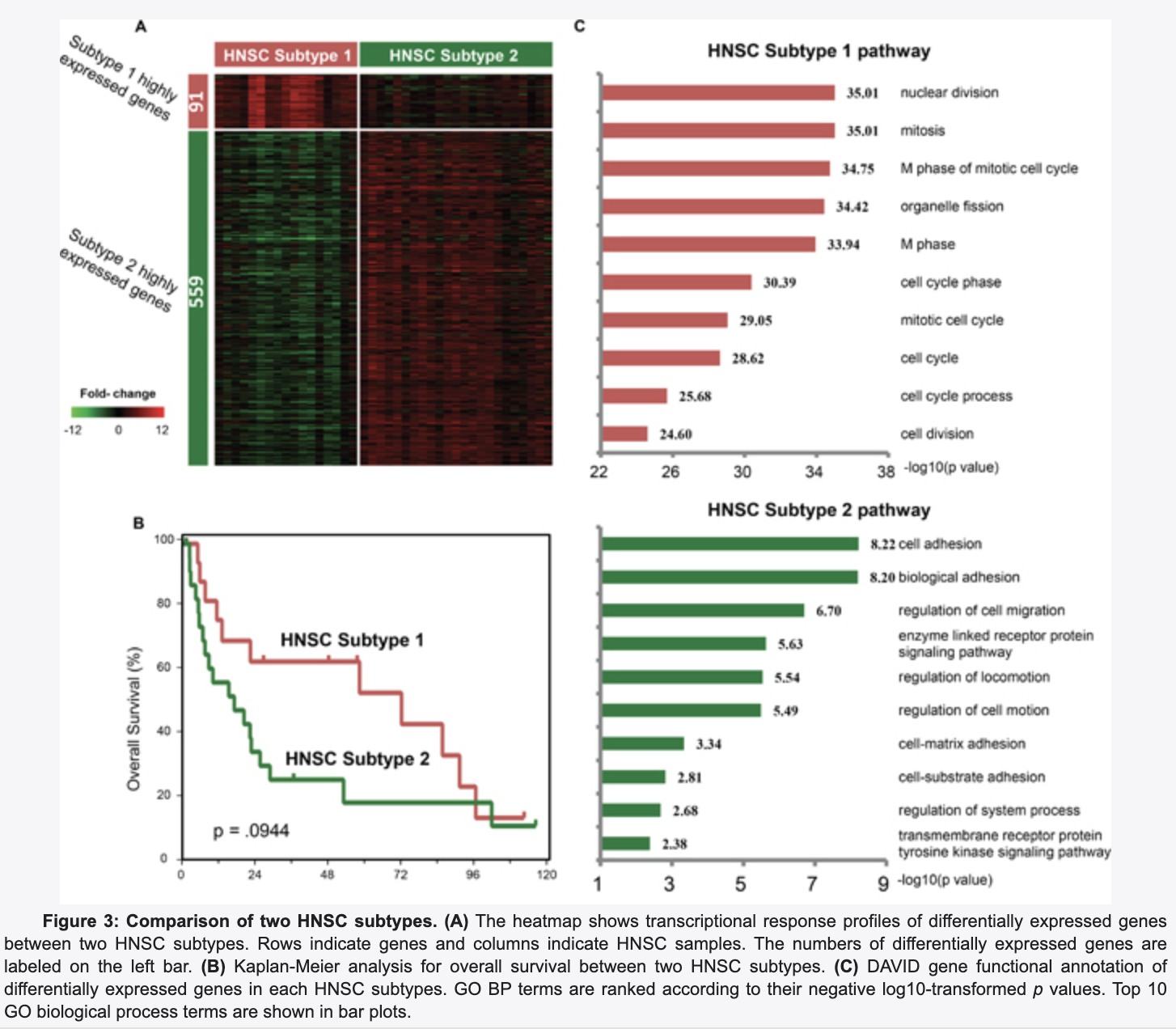
看完这个,你对单细胞数据分析是否有了基础认识了?如果你对单细胞数据分析还没有基础认知,可以看基础10讲: