Bioconductor系列包的安装方法都一样
source("http://bioconductor.org/biocLite.R")biocLite(“GenomicAlignments”)
这个包设计就是用来处理bam格式的比对结果的,具体功能非常多,其实还不如自己写脚本来做这些工作,更方便一点,实在没必要学别人的语法。大家有兴趣可以看GenomicAlignments的主页介绍,三个pdf文档来介绍。
http://www.bioconductor.org/packages/release/bioc/html/GenomicAlignments.html
我们首先读取示例pasillaBamSubset包带有的bam文件
library(pasillaBamSubset)
un1 <- untreated1_chr4() # single-end reads
library(GenomicAlignments)
reads1 <- readGAlignments(un1)
查看reads1这个结果,可以看到把这个bam文件都读成了一个数据对象GAlignments object,
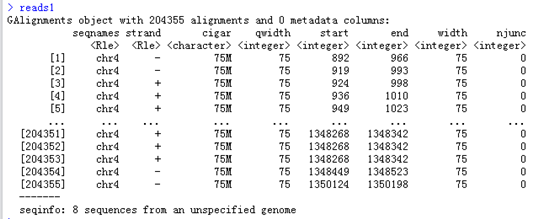
readGAlignments这个函数就是读取我们bam文件的,生成的对象可以用多个函数来继续查看信息,比如它的列名都是函数
Seqnames(),strand(),cigar(),qwidth(),start(),end(),width(),njunc() 这些函数对这个GAlignments对象处理后会得到比对的各个情况的向量。
我们也可以读取这个GenomicAlignments包自带的bam文件,而不是pasillaBamSubset包带有的bam文件
> fls <- list.files(system.file("extdata", package="GenomicAlignments"),+ recursive=TRUE, pattern="*bam$", full=TRUE)
> fls
[1] "C:/Program Files/R/R-3.1.2/library/GenomicAlignments/extdata/sm_treated1.bam" [2] "C:/Program Files/R/R-3.1.2/library/GenomicAlignments/extdata/sm_untreated1.bam"
reads1 <- readGAlignments(fls[1])
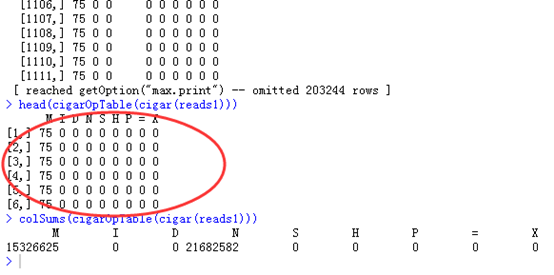
比如我随意截图 cigar(reads1)的结果,就可以看出,大部分都是75M这样的比对情况,可以对这个向量进行统计完全匹配的情况,总共有6077总比对情况。
unique(cigar(reads1))

也可以用table格式来查看cigar,可以看到大部分都是M
head(cigarOpTable(cigar(reads1)))
接下来我们再读取PE测序数据的比对bam结果,看看分析方法有哪些。

U3.galp <- readGAlignmentPairs(untreated3_chr4(), use.names=TRUE, param=param0)head(U3.galp)
也可以分开查看左右两端测序数据的比对情况,跟上面的head是对应关系,上面的
[169, 205] -- [ 326, 362]在下面就被分成左右两个比对情况了。
> head(first(U3.galp))
GAlignments object with 6 alignments and 0 metadata columns: seqnames strand cigar qwidth start end width njunc <Rle> <Rle> <character> <integer> <integer> <integer> <integer> <integer> [1] chr4 + 37M 37 169 205 37 0 [2] chr4 + 37M 37 943 979 37 0 [3] chr4 + 37M 37 944 980 37 0 [4] chr4 + 37M 37 946 982 37 0 [5] chr4 + 37M 37 966 1002 37 0 [6] chr4 + 37M 37 966 1002 37 0 ------- seqinfo: 8 sequences from an unspecified genome
> head(last(U3.galp))
GAlignments object with 6 alignments and 0 metadata columns: seqnames strand cigar qwidth start end width njunc <Rle> <Rle> <character> <integer> <integer> <integer> <integer> <integer> [1] chr4 - 37M 37 326 362 37 0 [2] chr4 - 37M 37 1086 1122 37 0 [3] chr4 - 37M 37 1119 1155 37 0 [4] chr4 - 37M 37 986 1022 37 0 [5] chr4 - 37M 37 1108 1144 37 0 [6] chr4 - 37M 37 1114 1150 37 0 ------- seqinfo: 8 sequences from an unspecified genome
用isProperPair可以查看pe测序数据比对情况的,哪些没有配对比对成功
> table(isProperPair(U3.galp))FALSE TRUE 29518 45828
还可以用Rsamtools包对我们的bam文件进行统计,看下面的代码中fls[1]和un1都是上面得到的bam文件全路径。
> library(Rsamtools)
> quickBamFlagSummary(fls[1], main.groups.only=TRUE)
group | nb of | nb of | mean / max of | records | unique | records per records | in group | QNAMEs | unique QNAMEAll records........................ A | 1800 | 1800 | 1 / 1 o template has single segment.... S | 1800 | 1800 | 1 / 1 o template has multiple segments. M | 0 | 0 | NA / NA - first segment.............. F | 0 | 0 | NA / NA - last segment............... L | 0 | 0 | NA / NA - other segment.............. O | 0 | 0 | NA / NA Note that (S, M) is a partitioning of A, and (F, L, O) is a partitioning of M.Indentation reflects this.
> library(Rsamtools)
> quickBamFlagSummary(un1, main.groups.only=TRUE)
group | nb of | nb of | mean / max of | records | unique | records per records | in group | QNAMEs | unique QNAMEAll records........................ A | 204355 | 190770 | 1.07 / 10 o template has single segment.... S | 204355 | 190770 | 1.07 / 10 o template has multiple segments. M | 0 | 0 | NA / NA - first segment.............. F | 0 | 0 | NA / NA - last segment............... L | 0 | 0 | NA / NA - other segment.............. O | 0 | 0 | NA / NA Note that (S, M) is a partitioning of A, and (F, L, O) is a partitioning of M.Indentation reflects this.
下面讲最后一个综合应用
biocLite("RNAseqData.HNRNPC.bam.chr14")
#这是一个665.5MB的RNA-seq测序数据的比对结果
library(RNAseqData.HNRNPC.bam.chr14)bamfiles <- RNAseqData.HNRNPC.bam.chr14_BAMFILESnames(bamfiles) # the names of the runslibrary(Rsamtools)quickBamFlagSummary(bamfiles[1], main.groups.only=TRUE)flag1 <- scanBamFlag(isFirstMateRead=TRUE, isSecondMateRead=FALSE, isDuplicate=FALSE, isNotPassingQualityControls=FALSE)param1 <- ScanBamParam(flag=flag1, what="seq") #这里是设置readGAlignments的读取参数library(GenomicAlignments)gal1 <- readGAlignments(bamfiles[1], use.names=TRUE, param=param1)
mcols(gal1)$seqoqseq1 <- mcols(gal1)$seqis_on_minus <- as.logical(strand(gal1) == "-")oqseq1[is_on_minus] <- reverseComplement(oqseq1[is_on_minus])
这样就还原得到了原始测序数据啦!