昨天在《生信技能树》公众号的一个教程:这也能画?,我提到了一个很无聊的R包,名字是:scRNAstat ,它可以4行代码进行单细胞转录组的降维聚类分群,其实完全没有技术含量, 就是把 Seurat 流程的一些步骤包装成为了4个函数:
- basic_qc (查看数据质量)
- basic_filter (进行一定程度的过滤)
- basic_workflow (降维聚类分群)
- basic_markers(检查各个亚群的标记基因)
我们这里以大名鼎鼎的pbmc3k数据集为例。如果你还没有下面的seurat-data包和pbmc3k对象 ,就自己去下载:
install.packages('devtools')
devtools::install_github('satijalab/seurat-data')
library(SeuratData) #加载seurat数据集
getOption('timeout')
options(timeout=10000)
InstallData("pbmc3k")
data("pbmc3k")
sce <- pbmc3k.final
这个时候,我们使用 pbmc3k数据集的这个sce变量,它是一个 Seurat 包需要的格式。然后我们创造一个文件夹来存放降维聚类分群结果,并且加载其它相关r包,主要是我的 scRNAstat :
library(scRNAstat)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(dplyr)
x='check_pbmc3k_by_scRNAstat'
dir.create( x )
接下来 就是正餐啦, 四行代码完成单细胞转录组的降维聚类分群
sce = basic_qc(sce=sce,org='human',
dir = x)
sce = basic_filter(sce)
sce = basic_workflow(sce,dir = x)
markers_figures <- basic_markers(sce,
org='human',
group='seurat_clusters',
dir = x)
有了这些, 就可以很容易去判断不同单细胞亚群的生物学名字啦!
p_umap = DimPlot(sce,reduction = 'umap',
group.by = 'seurat_clusters',
label.box = T, label = T,repel = T)
p_umap+markers_figures[[1]]
ggsave(paste0('umap_markers_for_',x,'.pdf'),width = 12)
如下所示:
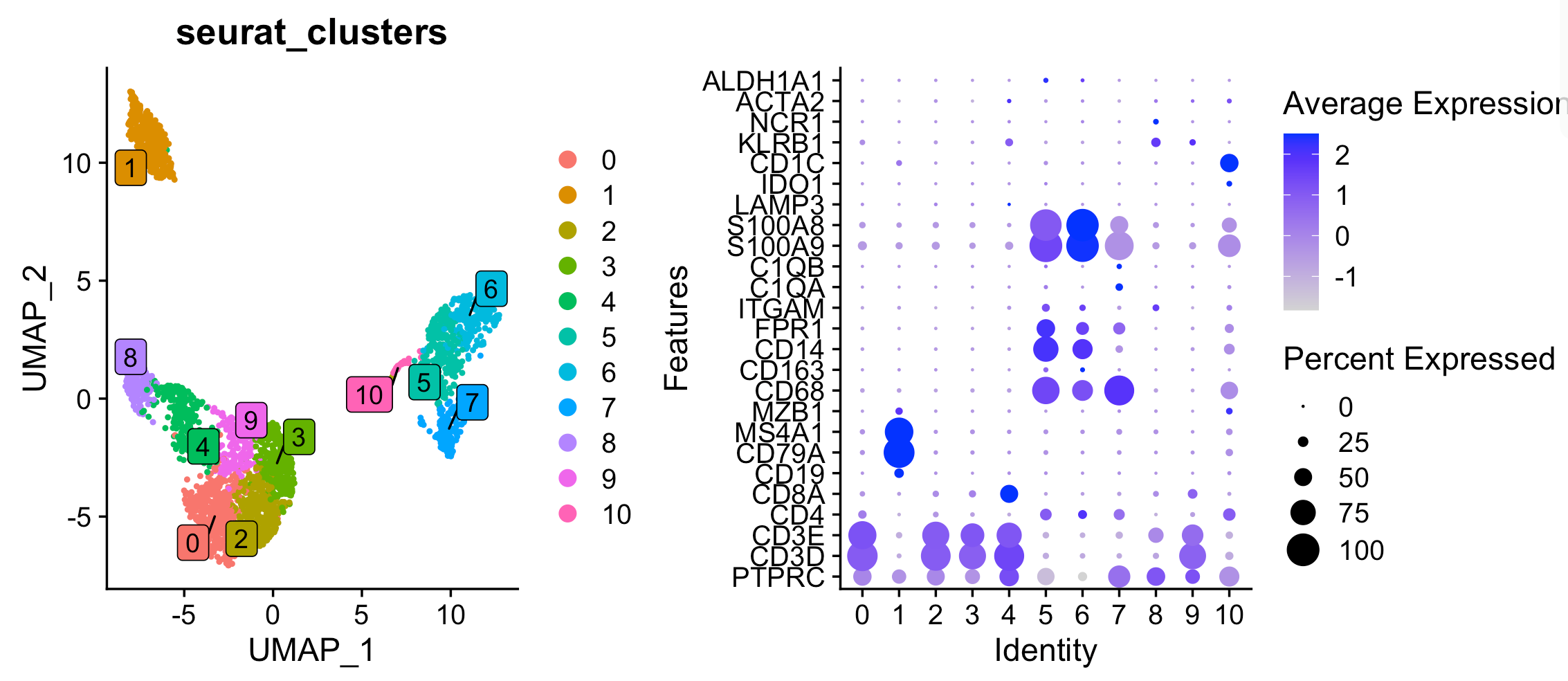
可以看到1群的B细胞,而4是CD8的T细胞,第8群是NK细胞,第10群的DC细胞,而0,2,3,9都是CD4的T细胞,其中5,6,7都是髓系而且主要是单核细胞。
给出这样的生物学名字,需要对上面的基因有一些背景知识哦!需要背诵如下所示各个细胞亚群高表达量基因的列表:
# T Cells (CD3D, CD3E, CD8A),
# B cells (CD19, CD79A, MS4A1 [CD20]),
# Plasma cells (IGHG1, MZB1, SDC1, CD79A),
# Monocytes and macrophages (CD68, CD163, CD14),
# NK Cells (FGFBP2, FCG3RA, CX3CR1),
# Photoreceptor cells (RCVRN),
# Fibroblasts (FGF7, MME),
# Endothelial cells (PECAM1, VWF).
# epi or tumor (EPCAM, KRT19, PROM1, ALDH1A1, CD24).
# immune (CD45+,PTPRC), epithelial/cancer (EpCAM+,EPCAM),
# stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
其实这样的基础认知,也可以看基础10讲:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
最基础的往往是降维聚类分群,参考前面的例子:人人都能学会的单细胞聚类分群注释
总结一下
对任意的 Seurat 包需要的变量格式,sce,都是可以走下面的 四行代码完成单细胞转录组的降维聚类分群:
sce = basic_qc(sce=sce,org='human',
dir = x)
sce = basic_filter(sce)
sce = basic_workflow(sce,dir = x)
markers_figures <- basic_markers(sce,
org='human',
group='seurat_clusters',
dir = x)
其实完全没有技术含量, 就是把 Seurat 流程的一些步骤包装成为了4个函数:
- basic_qc (查看数据质量)
- basic_filter (进行一定程度的过滤)
- basic_workflow (降维聚类分群)
- basic_markers(检查各个亚群的标记基因)