最近各个交流群总是看到大家询问一些单细胞公共数据集处理,居然是从bam文件开始,可能是因为都是从ENA数据库下载吧。
https://www.ebi.ac.uk/ena/browser/view/PRJNA578550?show=reads :
可以看到这个研究就两个样品,一个有fastq文件,一个是bam文件,非常的诡异,想下载其数据进行二次使用还很考验技术哦 :
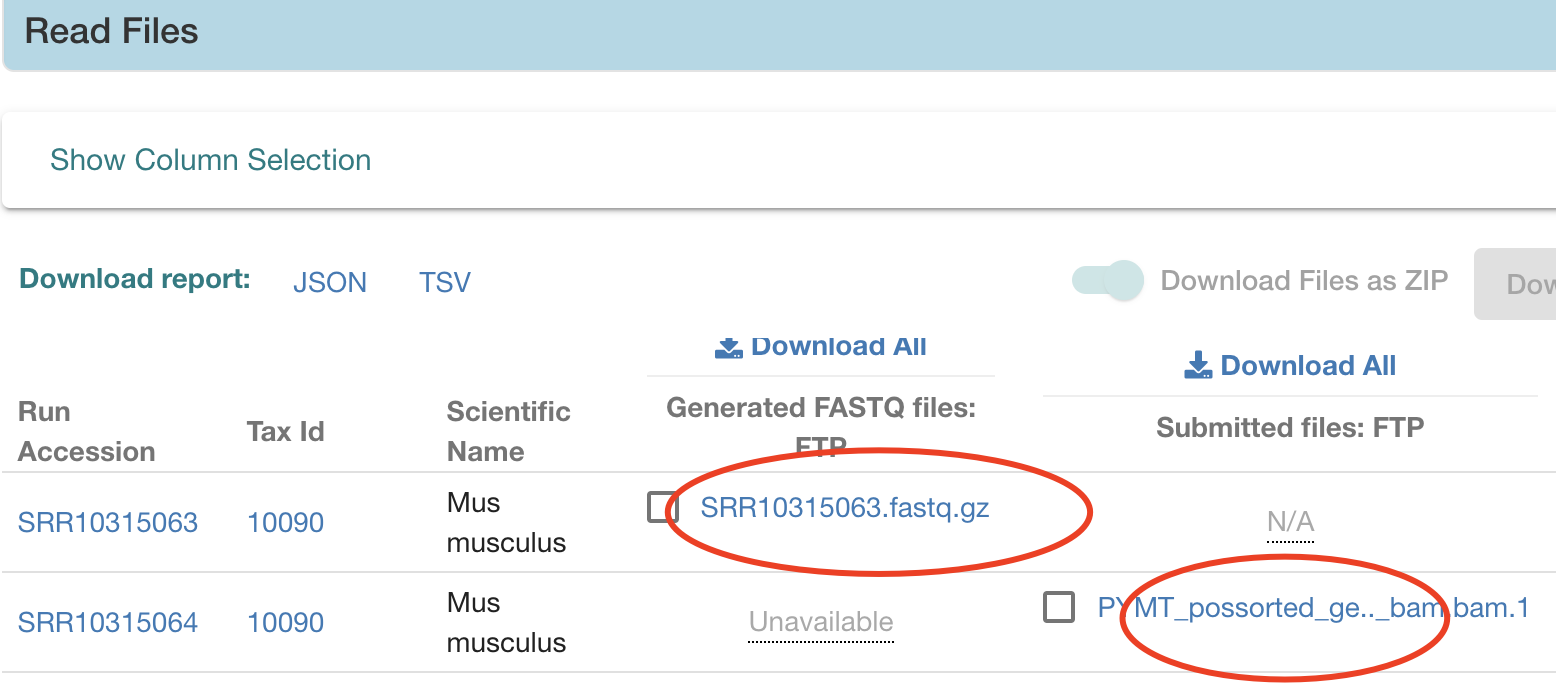
Droplet Based Single Cell RNA sequencing libraries were generated for 5 pooled FVB/n mouse spleens and 3 pooled PYMT mouse spleens using the 10x Genomics Chromium Platform and sequenced on the Illumina HighSeq 4000
其实这个bam文件就是标准的cellranger流程拿到的,其实目前单细胞转录组以10X公司为主流,我们也是在单细胞天地公众号详细介绍了cellranger全部使用细节及流程,大家可以自行前往学习,如下:
但是这个两年前的系列笔记是基于V2,V3版本的cellranger,在2020的7月我看到了其更新到了V4,也里面写了一个总结,见:cellranger更新到4啦(全新使用教程)
10x官网下载pbmc3k的bam文件
因为恰好最近也需要pbmc3k的bam文件做RNA速率分析,所以就选择10x官网下载pbmc3k的bam文件作为案例给大家演示。
链接在这 https://www.10xgenomics.com/resources/datasets/3-k-pbm-cs-from-a-healthy-donor-1-standard-1-1-0
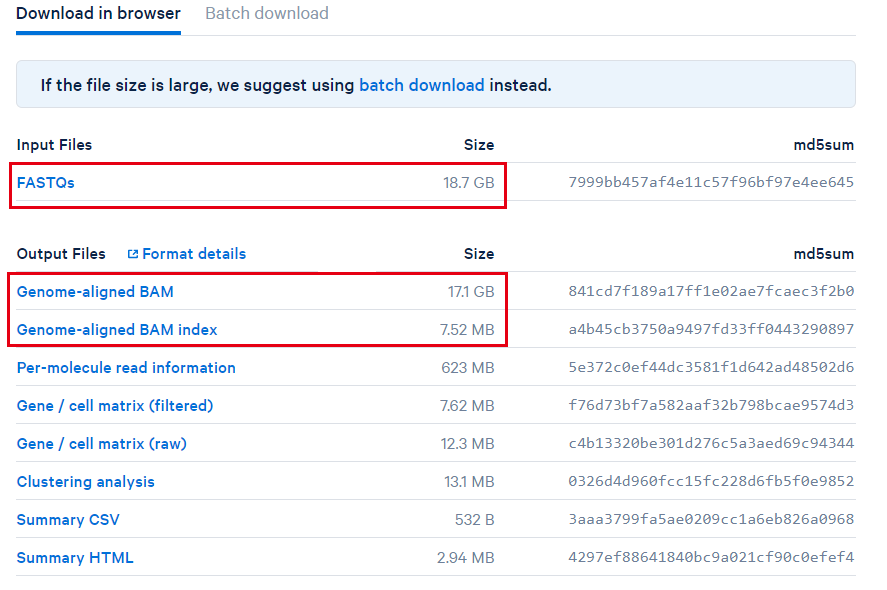
我们可以选择下载fq或者bam文件,由于pbmc3k是v1试剂时期的产物,他的fq文件比较古老。
其实10x官网的pbmc3k有fq文件,包含了多条lane,这里我们就不修改fq文件名了,因为可以直接下载bam文件,然后转换成fq的。
bamtofastq软件安装
首先我们要下载bamtofastq软件 , 根据自己的操作 系统选择版本。https://support.10xgenomics.com/docs/bamtofastq#header
wget -c https://github.com/10XGenomics/bamtofastq/releases/download/v1.4.1/bamtofastq_linux
chmod +x bamtofastq_linux
./bamtofastq_linux --help
# bamtofastq v1.4.1
# 10x Genomics BAM to FASTQ converter.
这个bamtofastq软件 小工具,参数还是蛮多的:
Usage: bamtofastq [options] <bam> <output-path> bamtofastq (-h | --help) Options: --nthreads=<n> Threads to use for reading BAM file [default: 4] --locus=<locus> Optional. Only include read pairs mapping to locus. Use chrom:start-end format. --reads-per-fastq=N Number of reads per FASTQ chunk [default: 50000000] --relaxed Skip unpaired or duplicated reads instead of throwing an error. --gemcode Convert a BAM produced from GemCode data (Longranger 1.0 - 1.3) --lr20 Convert a BAM produced by Longranger 2.0 --cr11 Convert a BAM produced by Cell Ranger 1.0-1.1 --bx-list=L Only include BX values listed in text file L. Requires BX-sorted and index BAM file (see Long Ranger support for details). --traceback Print full traceback if an error occurs. -h --help Show this screen.
接下来运行bamtofastq,需要注意的是 --cr11 参数哦,其实bam文件里面有其来源的软件版本和命令:
./bamtofastq_linux --cr11 pbmc3k_possorted_genome_bam.bam ./fastqs
得到fastq文件,如下所示:
tree fastqs/
fastqs/
└── gemgroup001
├── bamtofastq_S1_L000_I1_001.fastq.gz
├── bamtofastq_S1_L000_I1_002.fastq.gz
├── bamtofastq_S1_L000_I1_003.fastq.gz
├── bamtofastq_S1_L000_I1_004.fastq.gz
├── bamtofastq_S1_L000_R1_001.fastq.gz
├── bamtofastq_S1_L000_R1_002.fastq.gz
├── bamtofastq_S1_L000_R1_003.fastq.gz
├── bamtofastq_S1_L000_R1_004.fastq.gz
├── bamtofastq_S1_L000_R2_001.fastq.gz
├── bamtofastq_S1_L000_R2_002.fastq.gz
├── bamtofastq_S1_L000_R2_003.fastq.gz
├── bamtofastq_S1_L000_R2_004.fastq.gz
├── bamtofastq_S1_L000_R3_001.fastq.gz
├── bamtofastq_S1_L000_R3_002.fastq.gz
├── bamtofastq_S1_L000_R3_003.fastq.gz
└── bamtofastq_S1_L000_R3_004.fastq.gz
下面就可以基于上面的fastq文件,可以重新来运行 cellranger count了,也是超级简单 :
db=/home/bakdata/x10/pipeline/refdata-gex-GRCh38-2020-A
bin=/home/bakdata/x10/pipeline/cellranger-6.1.2/bin
$bin/cellranger count --id=pbmc3k \
--fastqs=./fastqs/gemgroup001 \
--transcriptome=$db \
--expect-cells=3000