前面我们通过生存分析聚焦到了恶性增殖的亚群,详见:关键单细胞亚群辅助判定之生存分析,而且也介绍了其实生存分析后可以做一个 最简单的统计学之取交集。
但是这两个方法的步骤以及代码量都很多,而且都是首先需要降维聚类分群后才能去确定具体的有意义的亚群。如果我们的降维聚类分群步骤里面的亚群并不纯粹或者分群时候的分辨率选择不够好,就会面临一个问题是得到的亚群就是混杂的,因为亚群是我们的最小分析单位。我们是依据每个单细胞亚群的特异性高表达量基因去给病人样品分组后生存分析,或者跟生存分析的基因取交集。
实际上,也是可以无需降维聚类分群,直接找生存相关的单细胞,就具体到每个细胞层面了,脱离了亚群的范畴。这里我们首先推荐Scissor,它发表在2021年Nature Biotechnology上,题为《Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data》 ,Scissor算法可利用单细胞数据和bulk RNA-seq数据及表型信息识别与疾病高度相关的细胞亚群。让我们来一起看看代码吧:
首先需要安装和加载它:
rm(list = ls())
library(Seurat)
library(preprocessCore)
# devtools::install_github('sunduanchen/Scissor')
# https://api.github.com/repos/sunduanchen/Scissor/tarball/HEAD
# devtools::install_local('~/Downloads/sunduanchen-Scissor-311560a.tar.gz')
library(Scissor)
第一步:拿到单细胞表达量矩阵对象
在:换一个分析策略会导致文章的全部论点都得推倒重来吗,我们把一个单细胞转录组数据集进行基础的降维聚类分群,并且针对里面的上皮细胞亚群进行细分亚群,而且在 肿瘤单细胞转录组拷贝数分析结果解读和应用我们根据拷贝数情况判断了其中0,1,6还有cycle是恶性细胞亚群。我们就是从这个上皮细胞细分亚群里面拿到的单细胞表达量矩阵对象哈,具体的代码和文件在:关键单细胞亚群辅助判定之生存分析可以拿到!!!
这里我们是基于R语言的Scissor算法实现,所以拿到的单细胞表达量矩阵对象是Seurat格式的:
load('scRNA_for_scAB_Scissor.Rdata')
scRNA
table(scRNA$celltype)
sce=CreateSeuratObject(
counts = scRNA@assays$RNA@counts,
meta.data = scRNA@meta.data
)
# 下面的 Scissor::Seurat_preprocessing 函数需要的仅仅是表达量矩阵
sc_dataset <- Scissor::Seurat_preprocessing(
scRNA@assays$RNA@counts , verbose = T)
sc_dataset$celltype = scRNA$celltype
DimPlot(sc_dataset, reduction = 'umap',
label = T, label.size = 3) +
DimPlot(sc_dataset, reduction = 'umap',
group.by = 'celltype', label = T, label.size = 3)
ggplot2::ggsave('umap_by_celltype_and_Scissor.pdf',width =8)
可以看到9个肺腺癌病人的上皮细胞被harmony整合后上皮细胞可以区分出来正常上皮细胞亚群以及一些未知的恶性肿瘤细胞亚群。
而且可以看到不同肿瘤病人上皮细胞harmony处理与否会导致病人个体差异被抹平如果不走harmony整合,我们就需要针对每个病人研究肿瘤内部异质性如果走harmony流程,就可以研究整体异质性。。。
下面是 harmony整合后上皮细胞 细分亚群情况:
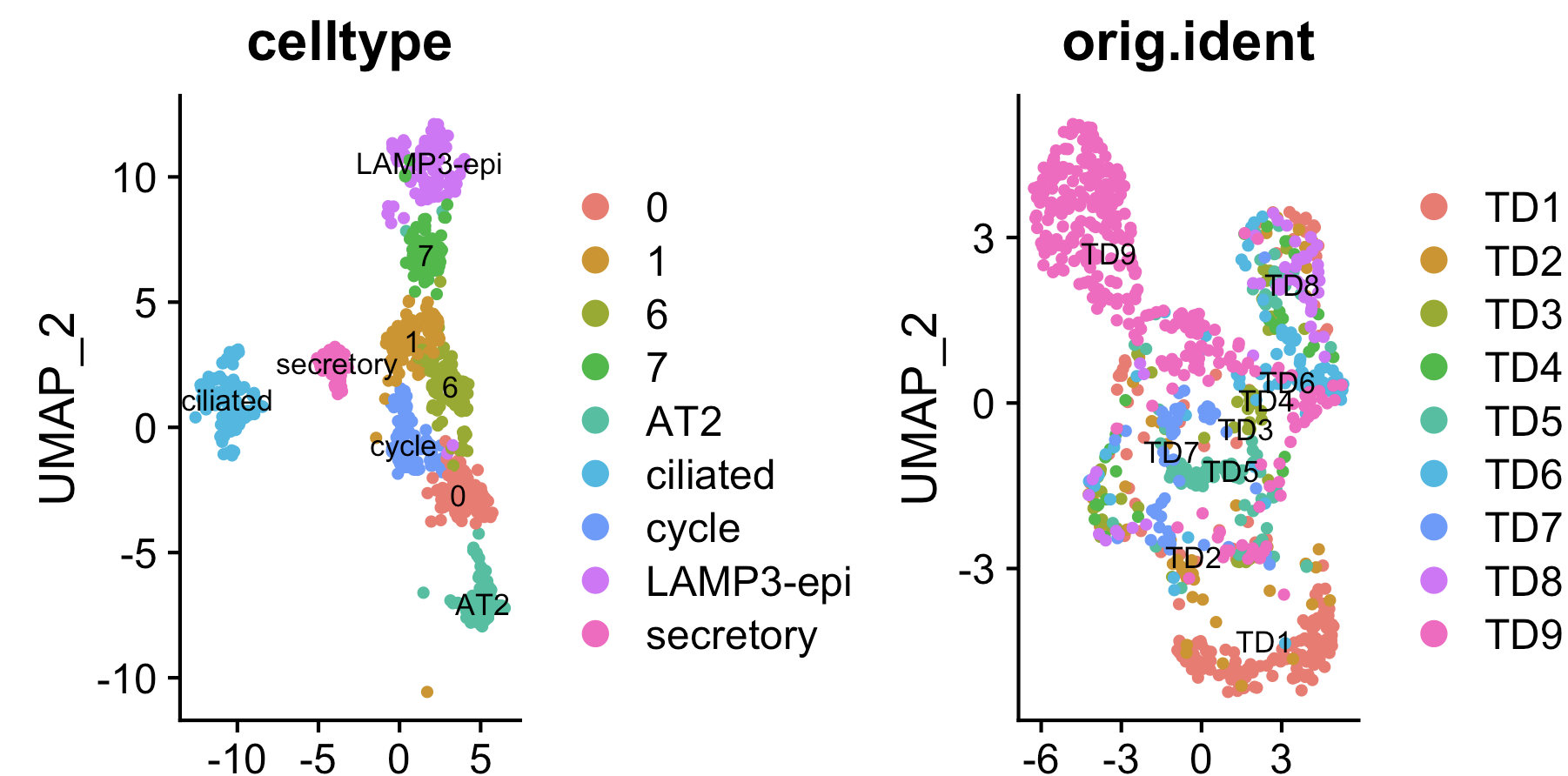
然后查看bulk转录组和病人临床信息:
这里就是前面的代码,具体的代码和文件在:关键单细胞亚群辅助判定之生存分析可以拿到!!!
load("../01-tcga_luad_from_xena/tcga-luad.for_survival.rdata")
head(meta)
exprSet[1:4,1:4]
bulk_dataset = exprSet
head(bulk_dataset[,1:10])
dim(bulk_dataset)
phenotype = meta[,c(3,2)]
colnames(phenotype)=c("time","status")
head(phenotype)
table(phenotype$status)
identical(colnames(bulk_dataset) ,row.names(phenotype))
一句话代码运行Scissor算法
非常奇怪的是我自己的Mac电脑居然运行半个小时以上,但是学徒的普通电脑就一两分钟。。。。
主要就是Scissor包Scissor函数而已,准备好单细胞对象,bulk表达量矩阵及其配对临床信息即可:
## 下面是主体代码,会很耗费时间和计算机资源
# 我的电脑超过了半个小时
if(T){
start_time1 <- Sys.time()
# devtools::install_github('sunduanchen/Scissor')
library(Seurat)
library(Scissor)
infos1 <- Scissor(as.matrix(bulk_dataset),
sc_dataset, phenotype, alpha = 0.05,
family = "cox",
Save_file = './Scissor_survival.RData')
end_time1 <- Sys.time()
execution_time1 <- end_time1 - start_time1
print(execution_time1)
}
names(infos1)
length(infos1$Scissor_pos)
infos1$Scissor_pos[1:4]
length(infos1$Scissor_neg)
infos1$Scissor_neg
可视化Scissor算法结果
Scissor_select <- rep("Background", ncol(sc_dataset))
names(Scissor_select) <- colnames(sc_dataset)
Scissor_select[infos1$Scissor_pos] <- "Scissor+"
Scissor_select[infos1$Scissor_neg] <- "Scissor-"
sc_dataset <- AddMetaData(sc_dataset,
metadata = Scissor_select,
col.name = "scissor")
table(sc_dataset$scissor)
p1 = DimPlot(sc_dataset, reduction = 'umap', group.by = 'scissor',
cols = c('grey','royalblue','indianred1'), pt.size = 1.2, order = T)
p1
p2 = DimPlot(sc_dataset, reduction = 'umap', group='celltype',
label = T, label.size = 3)
p2
p1+p2
gplots::balloonplot(
table(sc_dataset$celltype,sc_dataset$scissor)
)
save(infos1,sc_dataset,file = 'output_of_Scissor.Rdata')
很明显的可以看到这个Scissor算法判断的生存分析相关细胞主要是在恶性的增值细胞亚群:
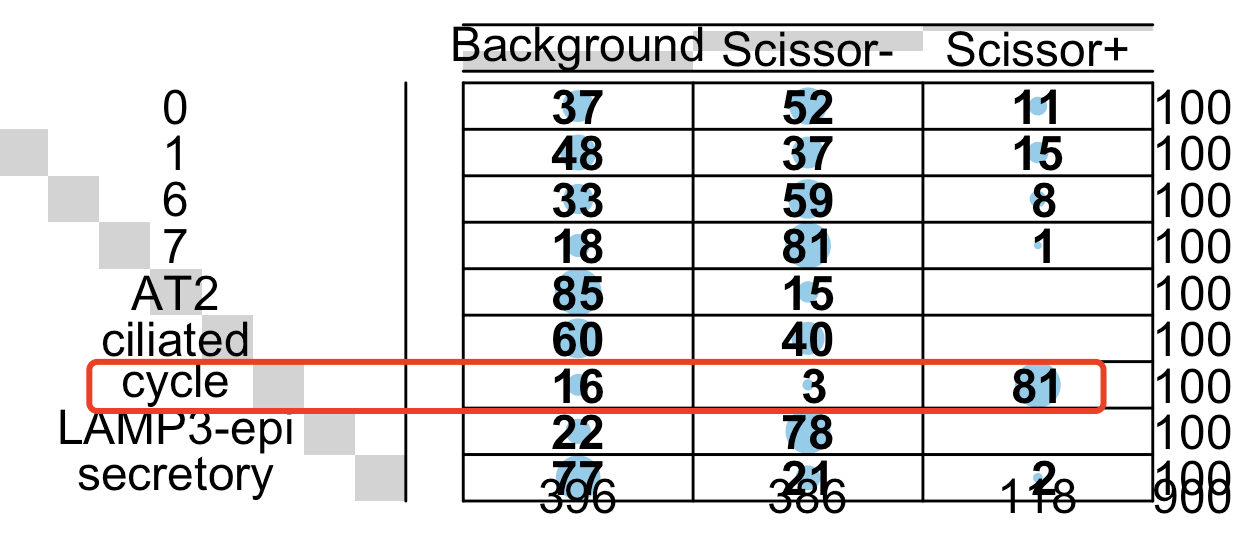
其中跟前面的:关键单细胞亚群辅助判定之生存分析,结论是一致的,但是前面是先降维聚类分群拿到了恶性的增值细胞亚群后根据生存分析结果判断恶性的增值细胞亚群的全部细胞都是有意义,但是Scissor算法判断的是恶性的增值细胞亚群绝大部分细胞是有意义的。
殊路同归,又有所不同!