学徒做了个GSE26305数据集的表达量芯片练习,总体上来说,经过了我们的标准分析训练,是可以完成差异分析和富集分析的,详见我十年前的公众号推文,目录在:
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
但是具体是到细节,学徒就傻眼了,比如文章里面提到了不同的样品使用这个芯片的时候仅仅是检测到1.33万个基因,如下所示:

人类的基因数量不止这一点,芯片设计也是覆盖全部的基因
2001 年,人类基因组计划宣布人类基因的初步估计为大约 20,000 到 25,000 个蛋白质编码基因。这个数字在当时引起了广泛的关注。然而,随着技术的进步和对基因组的更深入研究,人们逐渐认识到许多基因在不同条件下表现出多样性,一个基因可以产生多个不同的变体。此外,非编码RNA、微小RNA等功能元件的发现也扩展了对基因的理解。因此,现在的估计认为人类的蛋白质编码基因的数量可能比最初的估计要少。根据 GENCODE 项目,人类的蛋白质编码基因的数量约为 19,000 个,但这仍然是一个粗略的估计,因为不同的研究可能得出略有不同的数字。
首先根据gpl网页信息,我们可以得到探针对应的基因名字!
如下所示:
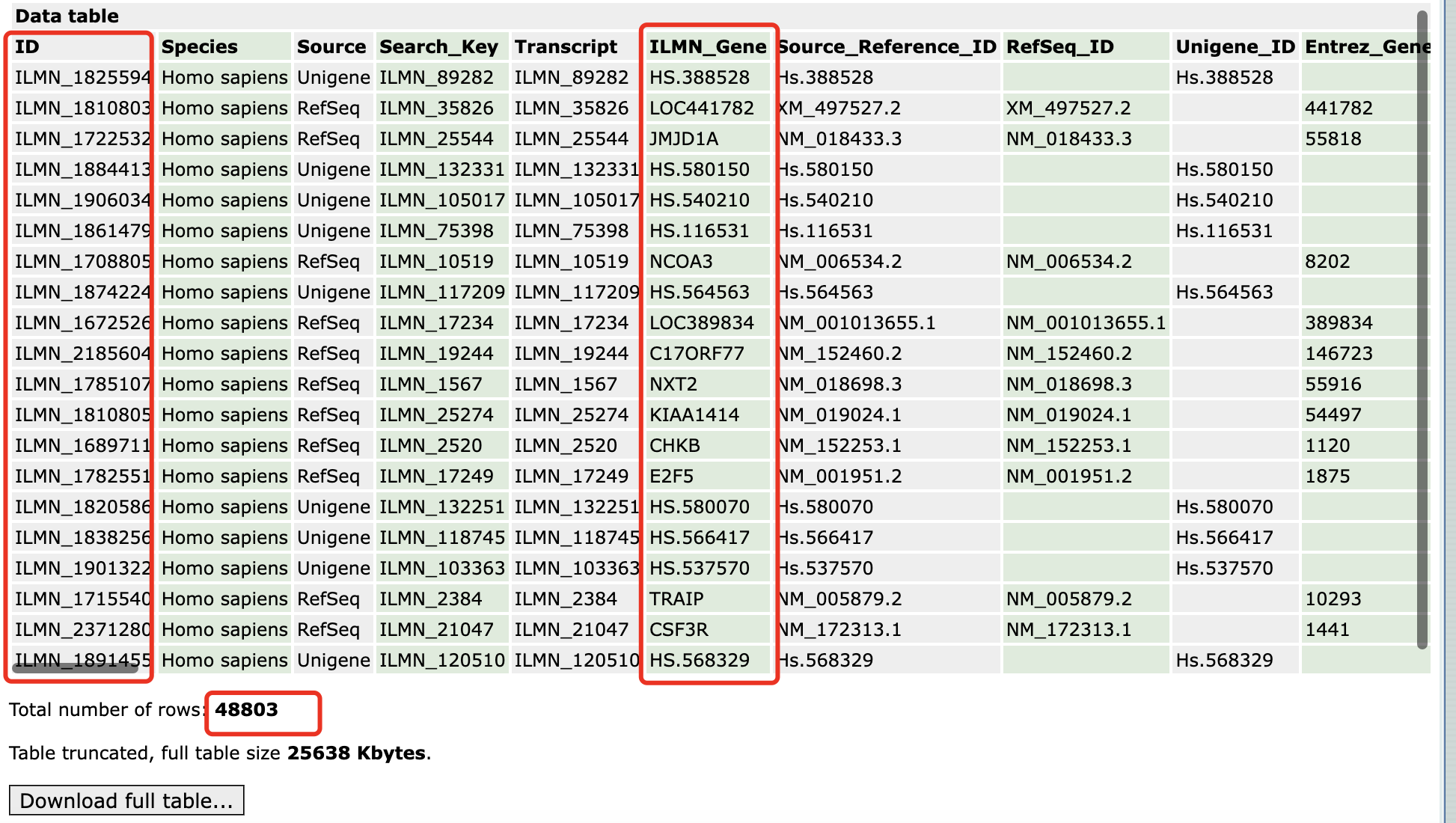
可以看到,起码有接近5万个探针,覆盖两万个左右的蛋白质编码基因是绰绰有余的。那么为什么文章里面提到了不同的样品使用这个芯片的时候仅仅是检测到1.33万个基因呢?
我们首先读取第一个文本文件,试试看:
library(data.table)
> fs=list.files('GSE26305_RAW/',full.names = T)
> fs
[1] "GSE26305_RAW//GSM645979.txt.gz" "GSE26305_RAW//GSM646008.txt.gz"
[3] "GSE26305_RAW//GSM646009.txt.gz" "GSE26305_RAW//GSM646010.txt.gz"
[5] "GSE26305_RAW//GSM646011.txt.gz" "GSE26305_RAW//GSM646012.txt.gz"
> a = fread(fs[1],data.table = F)
> head(a)
ID_REF Value Detection Pval
1 ILMN_1809034 111.5 0.00264
2 ILMN_1660305 334.7 0.00000
3 ILMN_1792173 210.9 0.00000
4 ILMN_1762337 46.0 0.88933
5 ILMN_2055271 57.2 0.11462
6 ILMN_1736007 46.0 0.88538
> table(a$`Detection Pval` < 0.05)
FALSE TRUE
29460 19343
可以看到, 每个样品有3列信息:
- 第一列是illumina的这个芯片的探针的id
- 第二列每个探针的表达量
- 第三列是每个探针的可靠性
然后我们问了问chatGPT,在Illumina的表达量芯片数据中,常见的几个列的含义如下:
- ID_REF (Probe ID): 这是每个探针的唯一标识符。每个基因在芯片上通常有一个或多个探针,而ID_REF列就是这些探针的标识。
- Value (表达量值): 这一列包含了探针的表达量值。这是一个连续的数值,表示该基因在样本中的表达水平。这个值可能经过一些预处理步骤,比如正则化或标准化。
- Detection (探测结果): Detection列通常包含有关每个基因是否检测到的信息。这个信息可能是基于阈值的二元结果,指示该基因是否在给定样本中被检测到。通常有 “P” 或者 “A” 表示检测到,”M” 表示边缘检测到,”NoCall” 表示未检测到。
- Pval (P值): 对于探测到的基因,Pval列通常包含与检测相关的统计学上的显著性水平,通常是一个P值。这个P值表示探针是否在该样本中显著地表达。在统计学中,P值越小,表明结果越显著。
总体而言,这些列提供了每个基因在每个样本中的表达量信息以及与其检测相关的统计学信息。在数据分析过程中,可以使用这些信息进行差异表达分析、可视化和其他生物信息学任务。
所以,我们会根据Detection Pval进行很简单的统计学显著性判断,就是p是否小于0.05来判断每个基因是否检测到的信息。然后这个信息就是基于阈值的二元结果,指示该基因是否在给定样本中被检测到。通常有 “P” 或者 “A” 表示检测到,”M” 表示边缘检测到,”NoCall” 表示未检测到。
我们简单的注释这些探针到基因上面:
load('GPL6884_soft.rda')
head(GPL6884_soft)
tmp = merge(a,GPL6884_soft,by.x = 'ID_REF',by.y='ID')
head(tmp)
length(unique(tmp[tmp$`Detection Pval` < 0.05,'symbol']))
可以看到探针信息合并了其基因注释信息后的能达到统计学显著的基因就是1.39万,跟原文是一模一样的 :
> head(tmp)
ID_REF Value Detection Pval symbol
1 ILMN_1343291 41281.9 0.00000 EEF1A1
2 ILMN_1343295 7879.8 0.00000 GAPDH
3 ILMN_1651199 52.5 0.31752 LOC643334
4 ILMN_1651209 51.3 0.40580 SLC35E2
5 ILMN_1651210 47.6 0.77734 DUSP22
6 ILMN_1651221 59.3 0.07510 LOC642820
> length(unique(tmp[tmp$`Detection Pval` < 0.05,'symbol']))
[1] 13930
也就是说,虽然说是芯片会覆盖两万个左右的蛋白质编码基因,但是很多探针可能是当次实验的信号值不够好,被pass了,这样的话,就只剩下了1.3万,也是很正常的。