看到了一个2023年11月发表的文章《Cross-talk between Myeloid and B Cells Shapes the Distinct Microenvironments of Primary and Secondary Liver Cancer》是非常简单的普通转录组结合单细胞转录组的数据分析案例,取样是 hepatocellular carcinoma (HCC) 和 colorectal cancer liver metastasis (CRLM)的两个分组,非常清晰的实验设计。
其中里面的普通转录组数据集链接是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE243245
可以看到,作者提供了这个转录组测序的表达量矩阵:GSE243245_RNA_seq_count.csv.gz 3.4 Mb
在R里面读入矩阵
代码很简单:
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
library(data.table)
rawcount <- fread("GSE243245_RNA_seq_count.csv.gz" ,data.table = F)
colnames(rawcount)
rawcount[1:4,1:4]
rownames(rawcount)=rawcount$V1
mat <-rawcount[,c(-1)]
mat[1:4,1:4]
rownames(mat)
keep_feature <- rowSums (mat > 1) > 1
table(keep_feature)
symbol_matrix <- mat[keep_feature, ]
group_list=ifelse(grepl('^m',colnames(symbol_matrix)),'case' ,'control')
table(group_list)
group_list
group_list = factor(group_list,levels = c('control','case' ))
save(symbol_matrix,group_list,file = 'symbol_matrix.Rdata')
很标准的矩阵:
> symbol_matrix[1:4,1:4]
HCC998T HCC2143T HCC2137T HCC2129T
A1BG 92543 4812 83067 357228
A1CF 12743 12152 5147 9418
A2M 18770 9962 202501 287747
A2ML1 3 2 0 0
因为作者给出来了的表达量矩阵:GSE243245_RNA_seq_count.csv.gz 3.4 Mb的列名非常有规律,我就简单的把 hepatocellular carcinoma (HCC) 和 colorectal cancer liver metastasis (CRLM)的两个分组,重新命名为了control和case组。
然后简单的质量控制,发现分组还是蛮合理的:
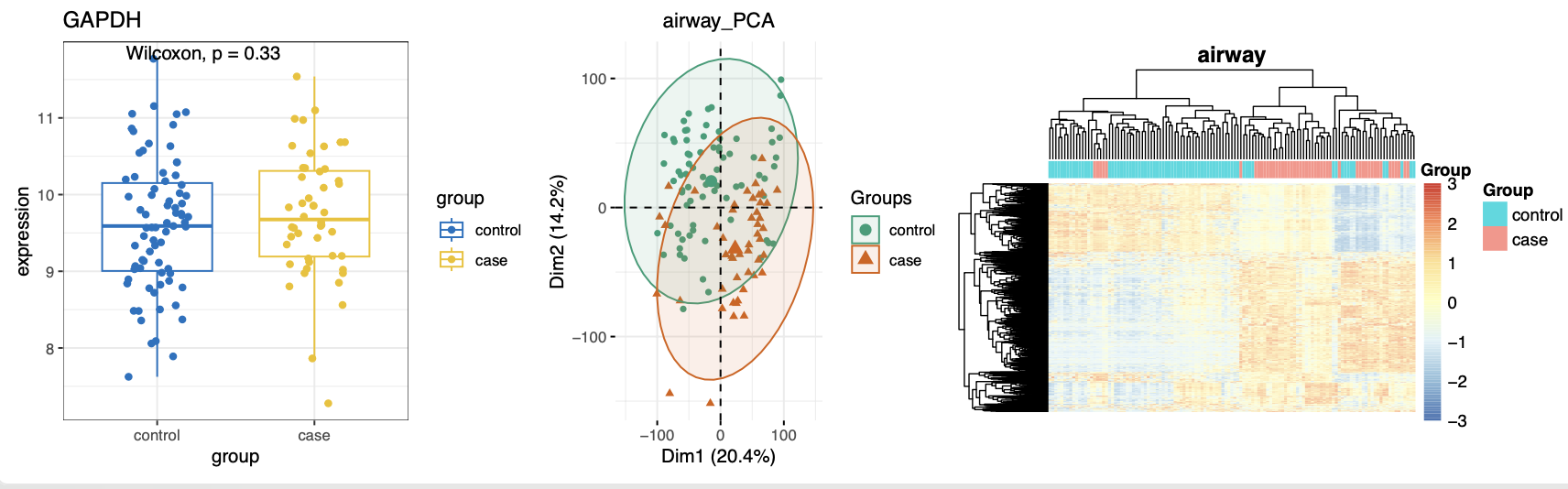
然后就是转录组测序的表达量矩阵的标准差异分析代码啦, 发现GAPDH基因作为内参确实是“稳如老狗”表达量是不会有差异变化,但是从差异基因居然可以倒推测出来其实是有一些control应该是case的,非常明显,如下所示 :
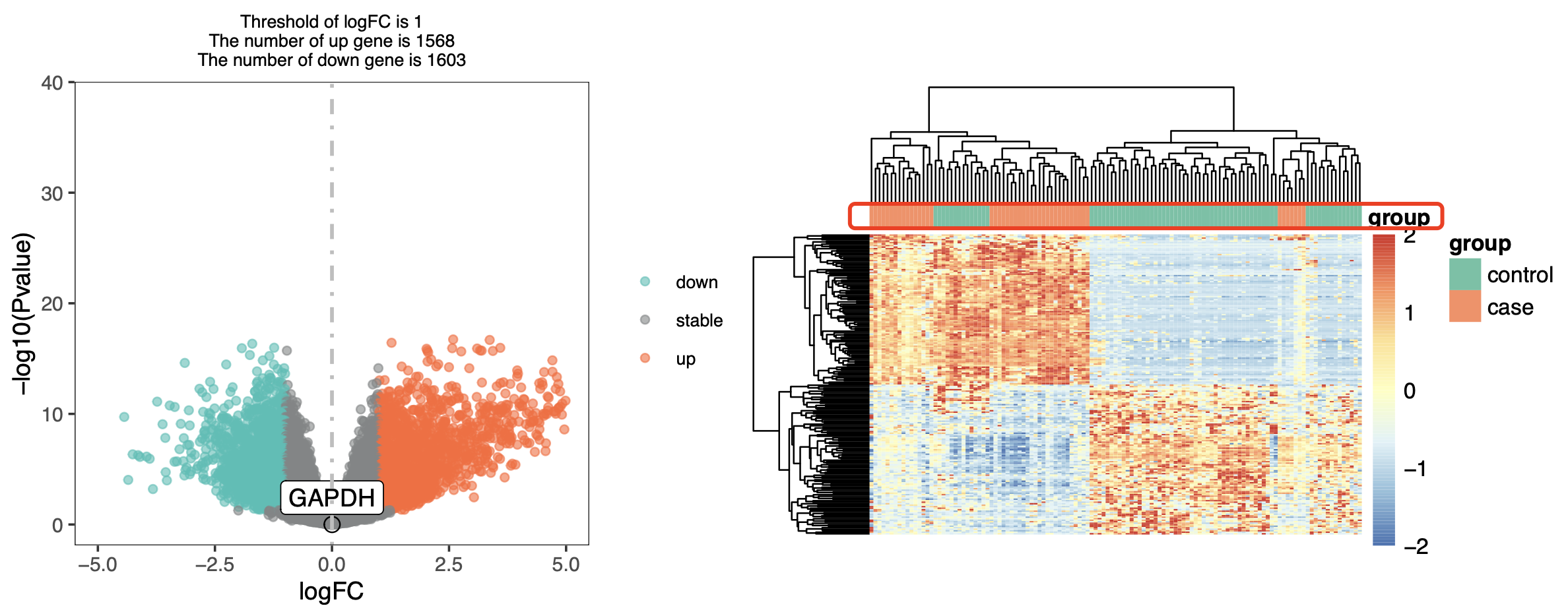
难道是可能是作者把部分样品标记错误了分组吗?
然后我仔细看了看文件名字
发现是我想简单了,以为是m开头的样品是colorectal cancer liver metastasis (CRLM),剩余的全部的hepatocellular carcinoma (HCC) :
> as.data.frame(table(gsub("[0-9]*","", colnames(symbol_matrix))))
Var1 Freq
1 HCC.P.T 1
2 HCCT 58
3 mCRC.S_T 5
4 mCRC.ST 43
5 SHCC.T 2
6 XLM 14
所以我做了一个简单的调整:
group_list=ifelse(grepl('^m',colnames(symbol_matrix)),'case' ,'control')
group_list=ifelse(grepl('^HCC',colnames(symbol_matrix)),'control','case' )
接下来出图就正常了:
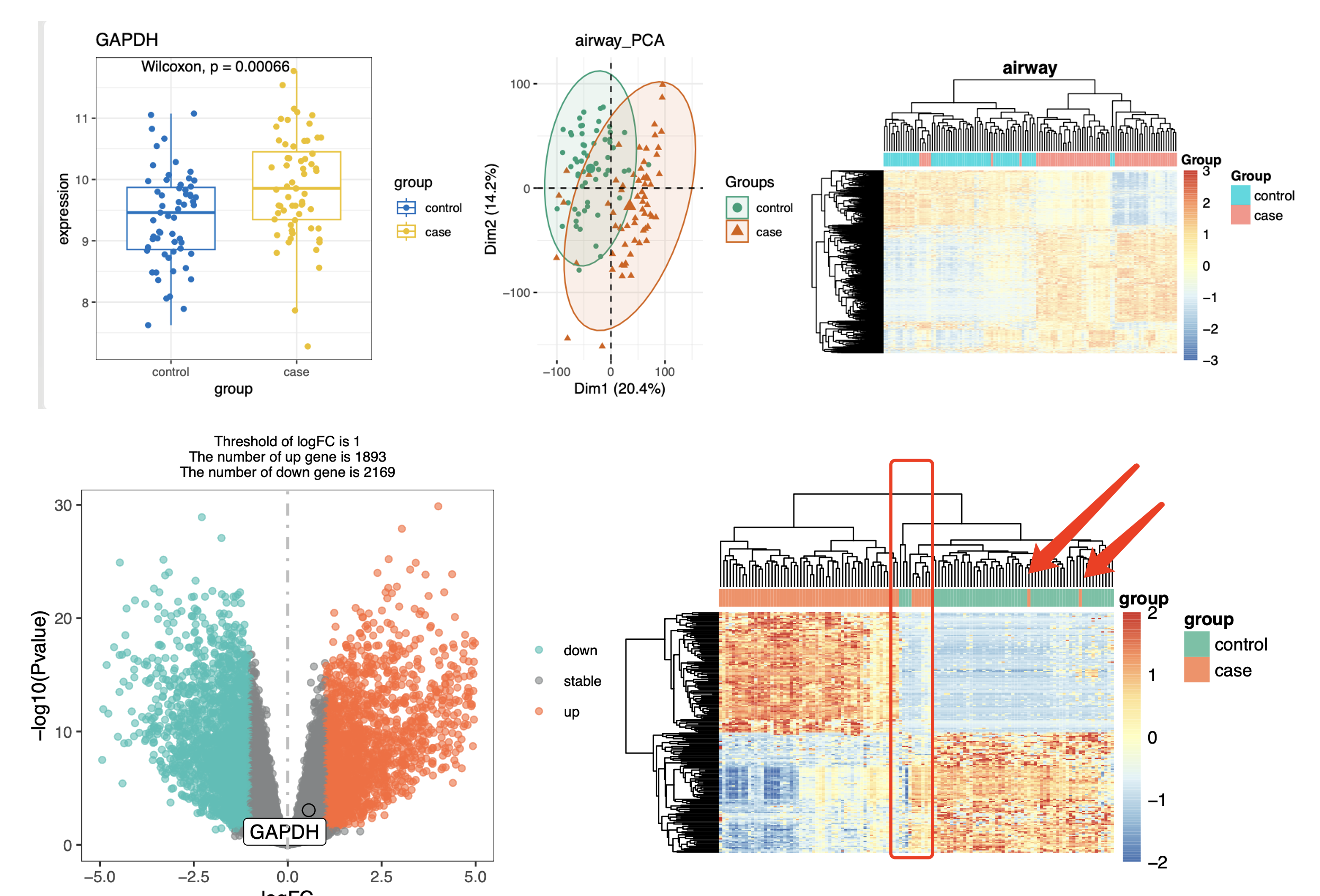
之前的一大批CRLM因为样品名字并不是以mCRC开头就被误标记为了HCC,这次被纠正过来了。
但是仍然是有一些样品是有hepatocellular carcinoma (HCC) 和 colorectal cancer liver metastasis (CRLM)的两个分组的混淆,而且有两个样品非常突兀,我用箭头高亮出来了。
学徒作业
上面的两次差异分析结果,大家可以做一个韦恩图,然后做一个全局散点图看看,最后说一下你的理解!
肝癌样品和结直肠癌的肝转移样品是有差异的
他们的差异这些差异涉及到细胞学、分子生物学、临床特征等多个层面。以下是可能存在的一些主要区别:
- 组织来源和病理特征:
- 肝癌样品通常是从原发于肝脏的恶性肿瘤中获取的,而结直肠癌的肝转移样品则来自原发于结肠或直肠的癌症,在肝脏发生了转移。
- 病理特征上,原发癌症和转移瘤可能在细胞形态、分化程度等方面存在不同。
- 分子生物学特征:
- 肝癌和结直肠癌在分子水平上可能有不同的遗传变异、突变谱、基因表达模式等。
- 转移瘤与原发瘤可能存在分子亚型的差异,包括在肿瘤抑制基因和促癌基因的表达上。
- 免疫组织化学标记:
- 肝癌和结直肠癌的肝转移样品可能在免疫组织化学标记上表现出不同的特征,例如免疫组织化学标记物(如肿瘤标记物)的表达水平。
- 病程特征:
- 肝癌和结直肠癌的肝转移可能在发病时机、生长速率、蔓延模式等方面存在差异。
- 肝转移可能对治疗的反应不同,包括对放疗、化疗、免疫治疗等的敏感性。
- 临床表现:
- 临床上,肝癌和结直肠癌的肝转移可能表现出不同的症状和体征,例如肝功能损害、腹痛、体重减轻等。
- 预后:
- 肝癌和结直肠癌的肝转移样品的预后可能存在差异,包括生存期、复发率等。
在转录组测序的信号层面可以达到百分百区分吗
基于上面的 hepatocellular carcinoma (HCC) 和 colorectal cancer liver metastasis (CRLM) 的两个分组的转录组测序的表达量矩阵很容易差异分析后,使用机器学习算法,比如LASSO,SVM, 随机森林缩小基因数量,来区分两个分组,调整算法和参数可以达到非常好的分类模型。
但是,它有没有临床价值呢?我们是否需要从转录组测序角度来区分hepatocellular carcinoma (HCC) 和 colorectal cancer liver metastasis (CRLM) 呢?
其次,因为首先我们的训练集里面可能会有标记错误的可能性(两个样品非常突兀,我用箭头高亮出来了),其次很明显会有少量样品是趋势层面就跟大部队格格不入,这样的话就不可能达到在转录组测序的信号层面可以达到百分百区分。