生物学功能注释是对特定的数量(几十个或者几百个)基因或蛋白的合集的功能进行描述和分类的过程。GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,科学家通常是使用来超几何分布检验这个统计学算法做富集分析,即通过比较实际观察到的基因集合(几十个或者几百个)中特定功能或通路的基因数量与随机期望的数量来判断其是否富集。
- 超几何分布检验的核心思想是,如果我们的基因集合(几十个或者几百个)是随机抽取的,那么它在某个特定功能类别(go以及kegg)中的富集情况应该是符合期望的。期望值是基于全部的基因(2万个左右)的分布来计算的。
- 比方说,我们差异分析或者其它分析拿到了200个基因,而全部的基因(是2万个左右),而且我们关注的某个特定功能类别(go以及kegg)在比如细胞增值这个通路是 100个基因。如果我们的抽取是随机的,我们预期在这个抽样中有多少基因属于我们关注的功能类别呢?
- 超几何分布检验会考虑这样一个问题:如果我们拿到了的200个基因里面有10个都是细胞增值这个通路里面的,概率如何?如果这种可能性很低,那么我们就认为我们观察到的富集情况是显著的,而不太可能是随机发生的。
如下所示的小洁老师授课PPT也分享了:
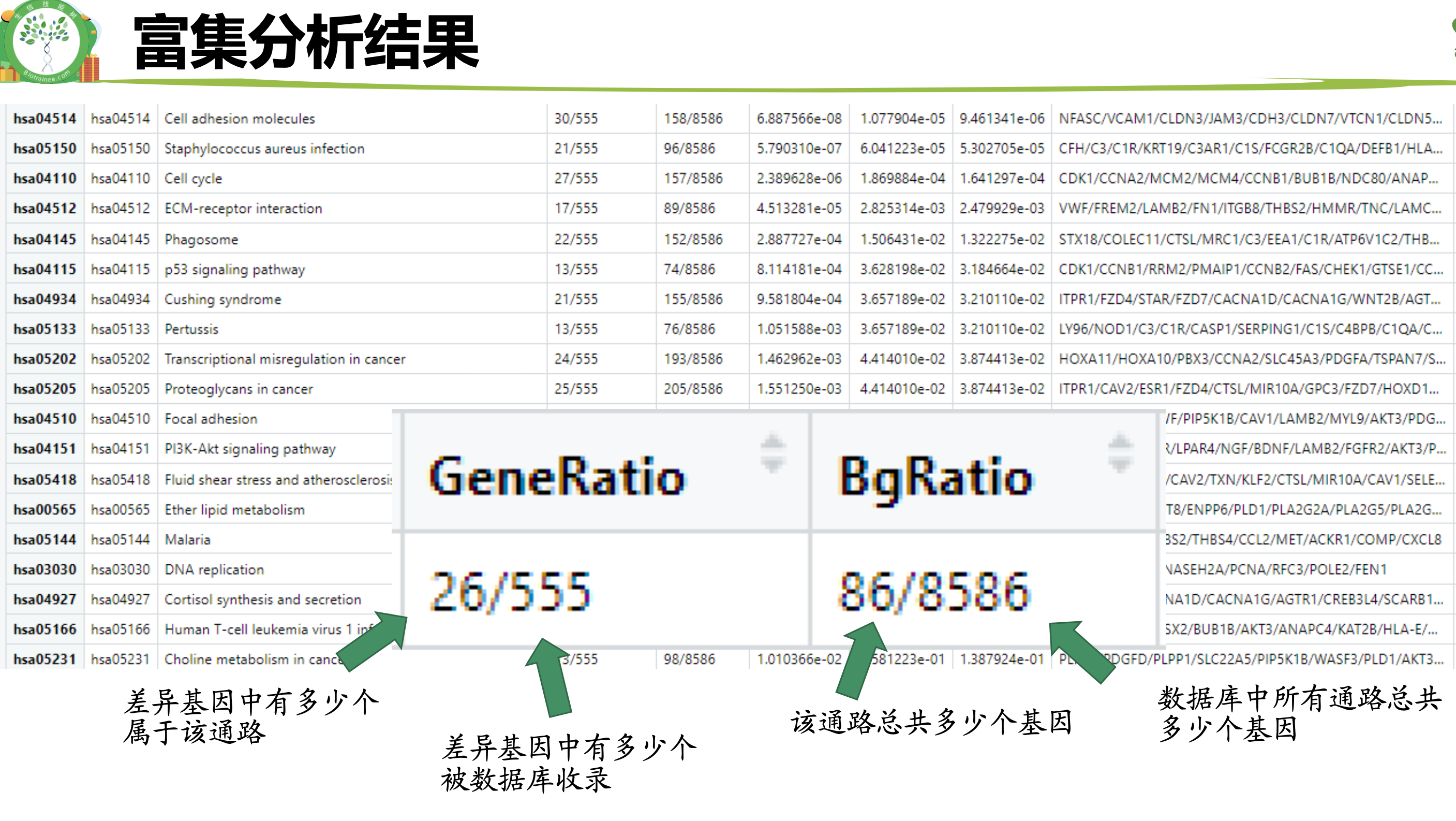
上面的案例里面的背景基因不到1万个,而差异基因是555个,有20倍的差距,理论上每个通路都是100左右数量级的基因理论上它们每个通路应该是就有5个左右的基因在差异基因列表里面。但是上面的通路的富集分析结果表格里面可以看到,绝大部分通路都是有十几个甚至二十多个基因在我们的差异基因列表里面,所以上面的通路都是被富集了。
最普通的就是go或者kegg数据库
其中GO注释通常包括三个方面的信息:分子功能(Molecular Function)、细胞组分(Cellular Component)和生物过程(Biological Process)。
如果大家对代码不熟悉,甚至可以使用在线网页工具直接完成生物学功能数据库(go以及kegg)注释(富集分析),最常见的是 3大在线分析工具:Enrichr、WebGestalt、gprofiler ,还有大名鼎鼎的DAVID(Database for Annotation, Visualization, and Integrated Discovery),以及后起之秀 Metascape,它们都可以用于功能富集分析的工具,帮助理解一组基因的生物学含义。
如果要更丰富就是MSigDB数据库啦
MSigDB(Molecular Signatures Database)数据库中定义了非常多的已知的基因集合,包括人类和小鼠的 :
需要简单的注册才能下载里面的gmt文件 ,其实就包含了前面提到的go和kegg数据库信息啦。这个gmt格式是broad研究所为他们开发的gsea分析定义的文本文件规范,就是每一行都是一个通路(基因集合),每个行所代表的通路可以是不限制的列。但是第一列必须是通路的名字或者ID,第二个是通路的描述,第三列以及之后的全部的列都是基因名字或者ID即可。

但是 msigdbr 这个R包 ,它内置了MSigDB(Molecular Signatures Database)数据库的全部基因集合。安装方法也非常简单:
install.packages("msigdbr")
包括H和C1-C8八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
- C8: cell type 相关的基因列表
有了 msigdbr 这个R包 ,就不需要去下载gmt文件进行解析啦,直接把 msigdbr 这个R包 里面的基因集合制作成为 GeneSetCollection 对象即可。
library(msigdbr)
all_gene_sets = msigdbr(species = "Homo sapiens",
category='H')
length(unique(table(all_gene_sets$gs_name)))
tail(table(all_gene_sets$gs_name))
gcSample = split(all_gene_sets$gene_symbol,
all_gene_sets$gs_name)
names(gcSample)
gs = lapply(gcSample, unique)
gsc <- GeneSetCollection(mapply(function(geneIds, keggId) {
GeneSet(geneIds, geneIdType=EntrezIdentifier(),
collectionType=KEGGCollection(keggId),
setName=keggId)
}, gs, names(gs)))
gsc
有了上面的这个gsc变量的 GeneSetCollection 对象 就可以很容易去做GSVA分析:
# 需要一个普通的表达量矩阵,X
es.max <- gsva(X, gsc,
mx.diff=FALSE, verbose=FALSE,
parallel.sz=8)
或者使用 clusterProfile 进行GSEA(GSEA analysis with clusterProfile) (前提是已获得排序好的genelist)(Assuming a well-range genelist ),这个时候不需要GeneSetCollection 对象 ,需要下载gmt文件,比如 h.all.v7.2.symbols.gmt文件,然后代码也是很简单:的
### 对 MsigDB中的全部基因集 做GSEA分析。
# http://www.bio-info-trainee.com/2105.html
# http://www.bio-info-trainee.com/2102.html
# https://www.gsea-msigdb.org/gsea/msigdb
# https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp
{
geneList= deg$avg_logFC
names(geneList)= toupper(rownames(deg))
geneList=sort(geneList,decreasing = T)
head(geneList)
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
#选择gmt文件(MigDB中的全部基因集)
gmtfile ='MSigDB/symbols/h.all.v7.2.symbols.gmt'
# 31120 个基因集
#GSEA分析
library(GSEABase) # BiocManager::install('GSEABase')
geneset <- clusterProfiler::read.gmt( gmtfile )
length(unique(geneset$term))
egmt <- GSEA(geneList, TERM2GENE=geneset,
minGSSize = 1,
pvalueCutoff = 0.99,
verbose=FALSE)
head(egmt)
egmt@result
gsea_results_df <- egmt@result
rownames(gsea_results_df)
write.csv(gsea_results_df,file = 'gsea_results_df.csv')
library(enrichplot)
gseaplot2(egmt,geneSetID = 'HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION',pvalue_table=T)
gseaplot2(egmt,geneSetID = 'HALLMARK_MTORC1_SIGNALING',pvalue_table=T)
}
更高级的是DoRothEA和PROGENy
- Discriminant Regulon Expression Analysis (DoRothEA)
- Pathway RespOnsive GENes for ac- tivity inference (PROGENy)
其实也可以仍然是理解为常规的gsea或者超几何分布检验,其中超几何分布检验是针对差异分析等拿到的基因集合(几十个或者几百个)进行生物学功能数据库注释,而常规的gsea或者gsva的分析是针对单个样品(需要有全部的两万多个基因)进行打分,所以会把表达量矩阵转为通路的打分矩阵。
而DoRothEA和PROGENy也是针对单个样品(需要有全部的两万多个基因)进行打分,可以把表达量矩阵转为转录因子或者肿瘤相关通路的打分,然后样品本身有分组就可以对前面的打分进行差异分析后展示。
让我们看看2023的文章《STK11/LKB1-Deficient Phenotype Rather Than Mutation Diminishes Immunotherapy Efficacy and Represents STING/Type I Interferon/CD8þ T-Cell Dysfunction in NSCLC》,是如何做这个分析的,如下所示:

就可以挑选有差异的地方去展示,如下所示:
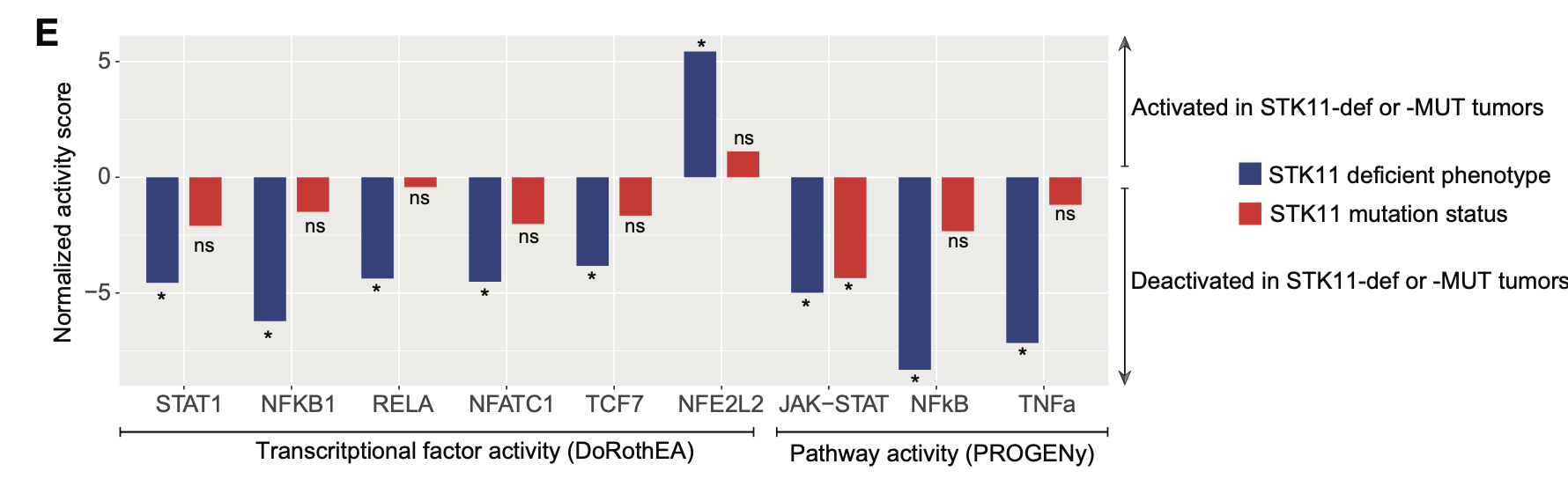
This notion was further supported by the DoRothEA and PROGENy analyses, which revealed that IFN-I–related and other immunostimulatory transcrip- tional factors (STAT1, NFKB1, RELA, NFATC1, TCF7, RELA) and pathways (JAK-STAT, NFkB, tumor necrosis factor-a) were significantly deactivated in STK11-def tumors,
学徒作业
下载airway这个R包后使用里面的表达量矩阵,然后进行DoRothEA和PROGENy这样的针对单个样品(需要有全部的两万多个基因)进行打分,可以把表达量矩阵转为转录因子或者肿瘤相关通路的打分,然后airway这个R包后使用里面的表达量矩阵的样品本身有分组就可以对前面的打分进行差异分析后展示。