前面我们在 初试Seurat的V5版本 的推文里面演示了文章标题是:《CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor》,的数据集GSE202642的Seurat的v5读取方式。
它虽然说是多样品,但是被作者整理成为了一个10x的样品的3文件格式, 所以很容易读取。接下来我们演示真正的Seurat的v5来读取多个10x的单细胞转录组矩阵。数据集在 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162616 可以看到作者给出来的矩阵还算是10X文件的3个标准文件,但是在每个样品下面都是3个文件,就是需要合理的修改文件名字而已:
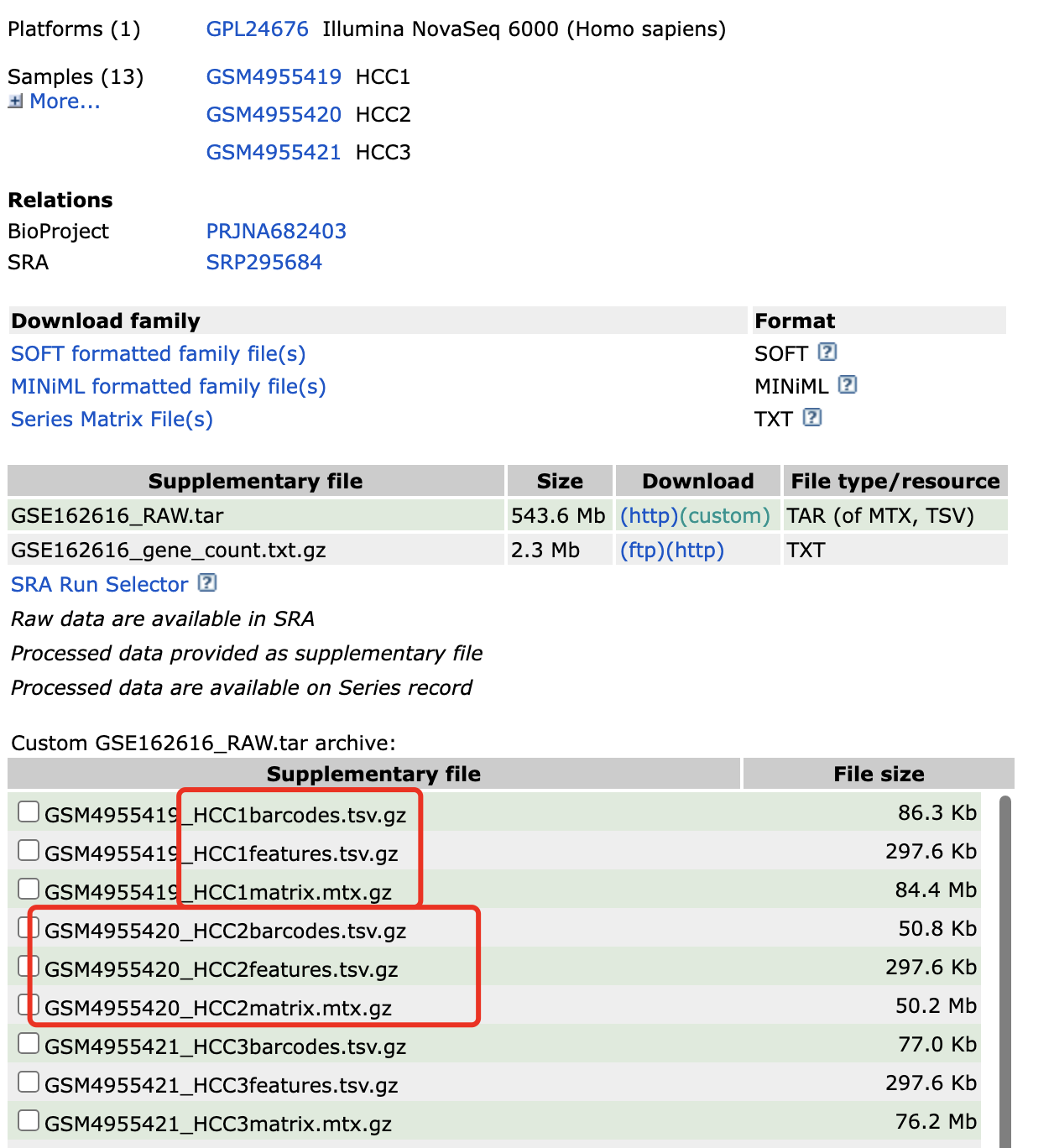
我们修改后是每个样品一个文件夹,每个文件夹里面的是10X文件的3个标准文件,如下所示:
$ tree -h GSE162616_RAW/outputs/
GSE162616_RAW/outputs/
|-- [ 0] HCC1
| |-- [ 86K] barcodes.tsv.gz
| |-- [298K] features.tsv.gz
| `-- [ 84M] matrix.mtx.gz
|-- [ 0] HCC2
| |-- [ 51K] barcodes.tsv.gz
| |-- [298K] features.tsv.gz
| `-- [ 50M] matrix.mtx.gz
`-- [ 0] HCC3
|-- [ 77K] barcodes.tsv.gz
|-- [298K] features.tsv.gz
`-- [ 76M] matrix.mtx.gz
3 directories, 9 files
如果我们按照之前Seurat的V4版本读取,代码如下所示:
dir='GSE162616_RAW/outputs/'
samples=list.files( dir )
samples
library(data.table)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
sce=CreateSeuratObject(counts = Read10X(file.path(dir,pro) ) ,
project = pro ,
min.cells = 5,
min.features = 300,)
return(sce)
})
names(sceList)
library(stringr)
# samples=gsub('.txt.gz','',str_split(samples,'_',simplify = T)[,2])
samples
names(sceList) = samples
sce.all <- merge(sceList[[1]], y= sceList[ -1 ] ,
add.cell.ids = samples)
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
# only the first layer is used
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
上面的代码我写了有好多年了,一直没有更新或者改进,我们依赖于这个V4的版本的Seurat流程做出来了大量的公共数据集的单细胞转录组降维聚类分群流程,100多个公共单细胞数据集全部的处理,链接:https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo但是最近其官方版本成为了V5……
因为现在是Seurat的V5版本,多个文件如果是分开读取后的merge函数其实并没有把每个样品的表达量矩阵merge,如下所示:
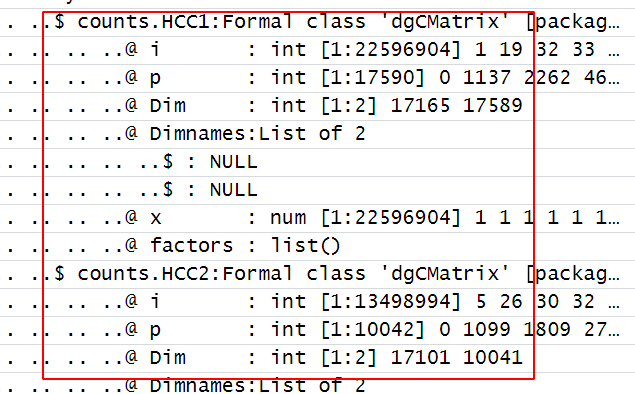
可以看到,在Seurat对象里面的每个样品仍然是独立的矩阵。。。。
这样的话,后面的流程就走不下去了,我们这个时候有一个很简单的方法就可以避免分开读取后的merge ,如下所示:
tmp = list.dirs('GSE162616_RAW/outputs/')[-1]
tmp
ct = Read10X(tmp)
sce.all=CreateSeuratObject(counts = ct ,
min.cells = 5,
min.features = 300,)
其实是因为这个函数Read10X可以一次性读取多个合理的路径,所以我们的如下所示的3个样品就被统一读取成为了一个稀疏矩阵而不是每个样品独立的稀疏矩阵,如下所示:
> tmp
[1] "GSE162616_RAW/outputs/HCC1"
[2] "GSE162616_RAW/outputs/HCC2"
[3] "GSE162616_RAW/outputs/HCC3"
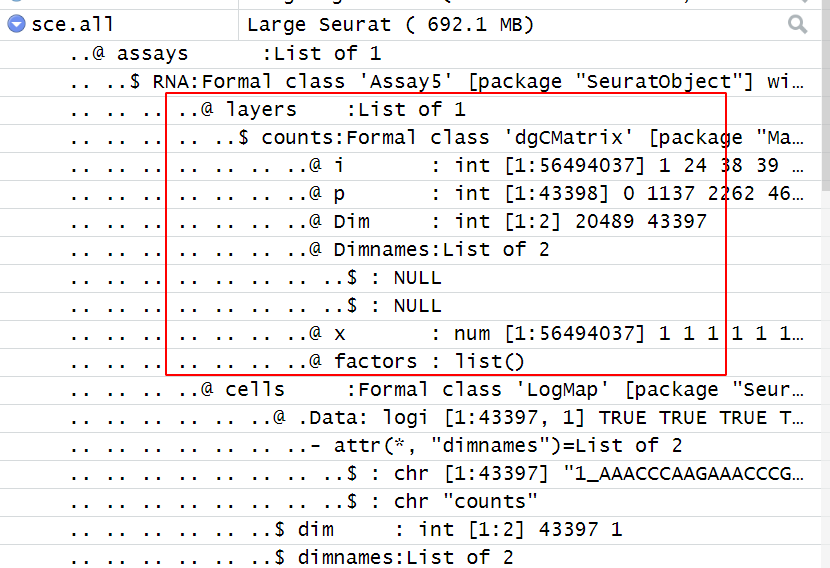
如果是对函数或者Seurat对象结构不清晰,就会产生如下所示错误的读取方式:
> sce.all=CreateSeuratObject(counts = Read10X('GSE162616_RAW/outputs/') ,
+ min.cells = 5,
+ min.features = 300,)
Error in Read10X("GSE162616_RAW/outputs/") :
Barcode file missing. Expecting barcodes.tsv.gz
这个 Read10X 函数能够接受一个或者多个合理的路径,合理的路径就是说里面有10X文件的3个标准文件,是不是很简单啊?
后面我们还会演示如何读取多个单细胞转录组样品,但是这些样品的矩阵并不是10x的3文件格式,所以会更麻烦一点!