马拉松授课的一个学员孜孜不倦的互动了十几个问题了,终于到了单细胞环节。
凭我对他的了解,他肯定是提问的方式就是错误的,写一段自己的”感悟“,其实完全没必要,我也压根不会看他给出来的这些“长篇大论” :
这样的提问完全没有用,没有代码,没有前因后果,其实给一下数据集就足够了,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145173
我们很容易自己去查看这个数据集的文件:
GSM4307836_SI_GA_H1_quants_mat.csv.gz 53.9 Mb
GSM4307836_SI_GA_H1_quants_mat_cols.txt.gz 430.6 Kb
GSM4307836_SI_GA_H1_quants_mat_rows.txt.gz 54.7 Kb
GSM4307837_SI_GA_H4_quants_mat.csv.gz 4.6 Mb
GSM4307837_SI_GA_H4_quants_mat_cols.txt.gz 430.6 Kb
GSM4307837_SI_GA_H4_quants_mat_rows.txt.gz 6.9 K
给人的感觉是,它这个文章作者对每个样品上传了3个文件,是很容易读取的。
GSM4307836 Mouse_PDGFRab_Sham
GSM4307837 Mouse_PDGFRab_UUO
但是实际上,作者给出来的 _quants_mat.csv.gz 并不是常规的表达量矩阵:
> library(data.table)
> a=fread('GSE145173_RAW/GSM4307836_SI_GA_H1_quants_mat.csv.gz',
+ data.table = F)
There were 50 or more warnings (use warnings() to see the first 50)
> a[1:4,1:4]
V1 V2 V3 V4
1 19 0 0 0
2 32 0 0 0
3 53 0 0 0
4 37 0 0 0
> dim(a)
[1] 10812 53699
如果是从这个行列式的数量来看,应该是1万多个细胞,然后是五万多个基因啦。
所以,如果是简单的基于这个 _quants_mat.csv.gz 文件去做单细胞转录组降维聚类分群是肯定是会有大麻烦!或者说, 如果是自己学艺不精,就会以为作者上传了错误的矩阵。因为最后这个读取确实是太复杂了:
{
library(data.table)
a=fread('GSE145173_RAW/GSM4307836_SI_GA_H1_quants_mat.csv.gz',
data.table = F)
head(a[,1:4])
tail(a[,1:4])
head(t(a)[,1:4])
tail(t(a)[,1:4])
dim(a)
cl=fread('GSE145173_RAW/GSM4307836_SI_GA_H1_quants_mat_cols.txt.gz',
header = F,data.table = F )
dim(cl)
length(unique(cl$V2))
kp = duplicated(cl$V2)
table(kp)
cl=cl[!kp,]
# 不知道为什么表达量矩阵跟它给出来的基因名字,行数不匹配,我被迫删除了其中两个基因,但是不知道是否造成了基因错位。。。。
a=a[,1:(ncol(a)-2)]
rl=fread('GSE145173_RAW/GSM4307836_SI_GA_H1_quants_mat_rows.txt.gz',
header = F,data.table = F )
head(rl)
mtx=t(a[,!kp])
dim(mtx)
rownames(mtx)=cl$V2
colnames(mtx)=rl$V1
mtx[1:4,1:4]
sce1=CreateSeuratObject(counts = mtx,project = 'h1' )
sce1
}
但是接下来我检查数据分析结果的时候,发现确实是造成了基因错位。。。。
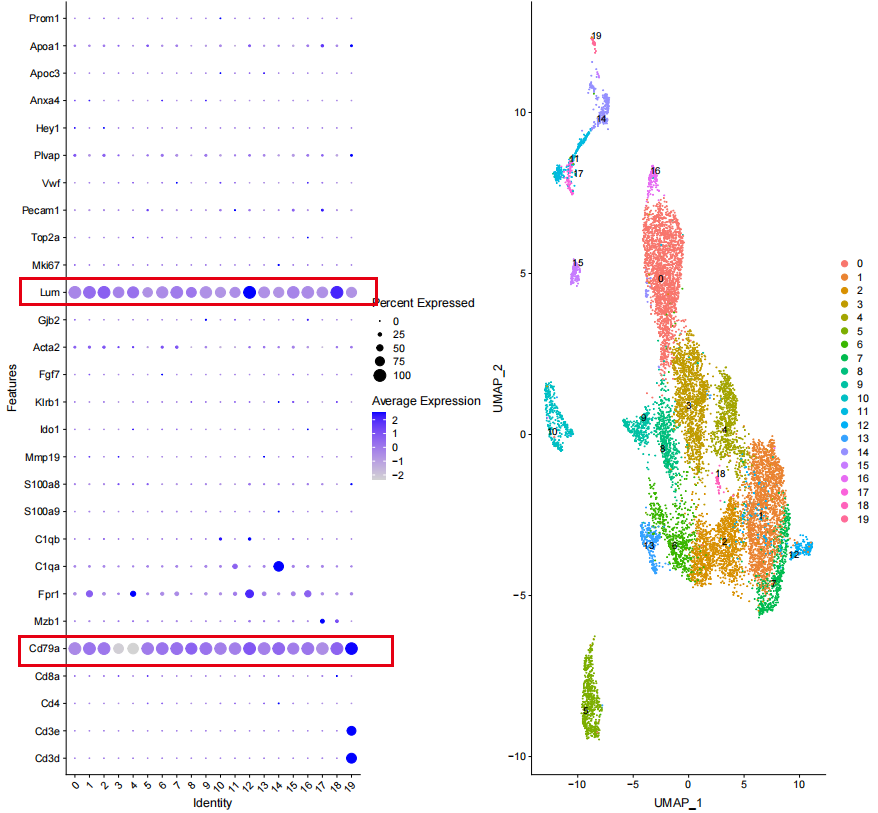
因为后面的降维聚类分群结果问题不大,但是基因在上面就显得很突兀,基本上没有任何一个我认识的基因。。。
Decoding myofibroblast origins in human kidney fibrosis. Nature 2021 Jan 人家的文章发表在CNS啊!
我实在是没办法理解, 既然同学们要重复使用他们的数据,居然不认真彻底读懂文章,简直是对科研的侮辱!!!但凡看了看文章就知道这个样品:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM4307836
给出来的是:raw UMI barcode count matrices produced by Alevin in csv files and corresponding row (gene) and column (cell barcode) information
虽然说这个单细胞确实是 10x chromium version 2 ,但是作者,走的是另外一个流程,Data processing Fastq files were processed using Alevin and Salmon (salmon version 0.13.1)
所以给出来的表达量矩阵格式是需要重新学习的,不能说仅仅是照搬10x的cellranger那一套!!!
官方文档写的很清楚:https://salmon.readthedocs.io/en/latest/alevin.html#
A typical run of alevin will generate 4 files:
- quants_mat.gz – Compressed count matrix.
- quants_mat_cols.txt – Column Header (Gene-ids) of the matrix.
- quants_mat_rows.txt – Row Index (CB-ids) of the matrix.
- quants_tier_mat.gz – Tier categorization of the matrix.
而且文章是给出来了全部的代码:
- All R and Python packages used in data analysis are described here: https://raw.githubusercontent.com/mahmoudibrahim/KidneyMap/master/requirements.txt. Additional software used for data analysis: Graphpad, FlowJo, ImageJ.
- All scripts used for single cell RNA-seq data analysis are available here: https://github.com/mahmoudibrahim/KidneyMap/
- Scripts used imaging in situ hybridization are available here: https://gitlab.com/mklaus/segment_cells_register_marker
直播预告
既然是上面的文章的数据可以使用 Alevin and Salmon ,那么理论上也可以使用10x的cellranger流程,我们就可以对比下了。我们在最新的唐医生服务器上面给大家演示吧!