最近单细胞数据挖掘文章如雨后春笋般冒出来了,总体来说就两个方向:
- 找到一个数据集,降维聚类分群后,拿到基因列表后,去TCGA建模
- 首先TCGA建模然后拿模型里面的基因去单细胞转录组数据集看是否有特殊的表现
这两个方向都需要掌握基础的单细胞转录组数据集的降维聚类分群,如果这个环节有问题就会造成数据挖掘文章很尴尬,比如:2023的文章:《 Integrated analysis of single‐cell and bulk RNA‐sequencing identifies a signature based on NK cell marker genes to predict prognosis and immunotherapy response in hepatocellular carcinoma 》,下载了数据集GSE162616里面的3个样品的单细胞数据,数据分析其实中规中矩,使用我们给大家的示范代码,三五分钟就可以出结果,但是我看到了它里面的NK细胞数量非常多,不太符合基础认知,如下所示:
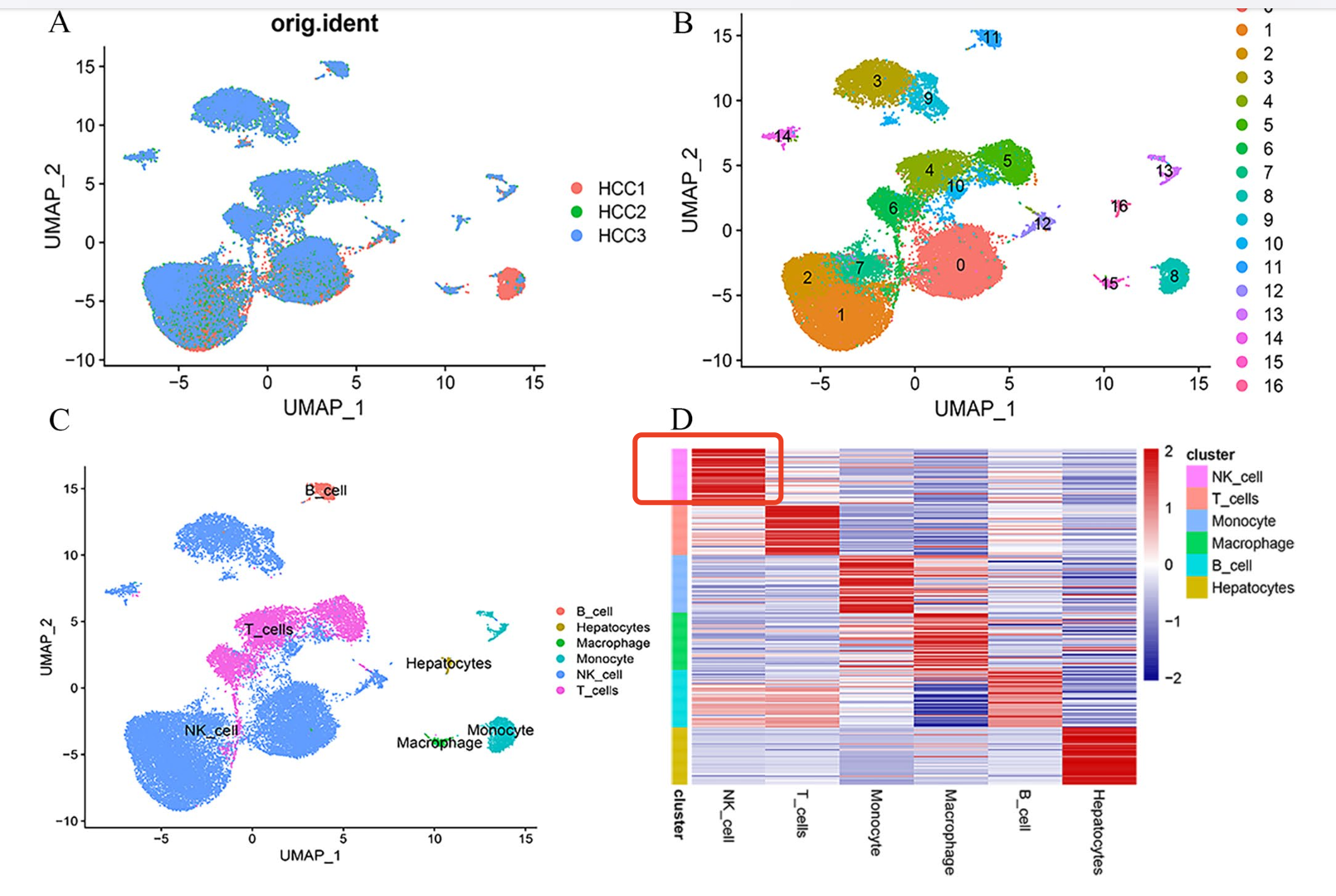
我看了看文章里面的描述;
- Cells in clusters 0, 1, 2, 3, 7, 9, 10, 12, and 14 were annotated as NK cells based on SingleR
问题就出在这里,SingleR并不是不能使用,毕竟也有那么多人引用,但是它很大概率会被误用,而且我们也不推荐它作为唯一的或者说是最后的单细胞亚群命名标准。
学徒作业
进入数据集:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162616
它里面有13个样品:
GSM4955419 HCC1
GSM4955420 HCC2
GSM4955421 HCC3
GSM4955422 HD1
GSM4955423 HD2
GSM4955424 HD3
GSM4955425 PT
GSM4955426 IT
GSM4955427 PBMC
GSM4955428 IP03
GSM4955429 IP18
GSM4955430 IN30
GSM4955431 IN03
需要大家定位到里面的3个HCC的单细胞样品的文件:
GSM4955419_HCC1barcodes.tsv.gz 86.3 Kb
GSM4955419_HCC1features.tsv.gz 297.6 Kb
GSM4955419_HCC1matrix.mtx.gz 84.4 Mb
GSM4955420_HCC2barcodes.tsv.gz 50.8 Kb
GSM4955420_HCC2features.tsv.gz 297.6 Kb
GSM4955420_HCC2matrix.mtx.gz 50.2 Mb
GSM4955421_HCC3barcodes.tsv.gz 77.0 Kb
GSM4955421_HCC3features.tsv.gz 297.6 Kb
GSM4955421_HCC3matrix.mtx.gz 76.2 Mb
读取它们,走降维聚类分群,然后看看亚群命名,真的是有这么多NK细胞吗?