最近接到了还没有入门的备选学徒的提问,发现其实可以使用chatGPT很方便的解决,如果大家不能使用外网的chatGPT,也可以参考我们通过BioinfoArk提供的中国区chatGPT的查询方式:
当然了,如何提问,就需要一点点背景知识啦, 比如知道什么是变异位点,什么是过滤,然后就可以很简单的两个提问即可:
- 先让chatGPT介绍一下bcftools工具,来龙去脉了解一下
- 然后让chatGPT细致的讲一下bcftools工具过滤变异的功能单元即可
“bcftools”(Binary Call Format tools)是一个用于处理Variant Call Format (VCF) 和 Binary Call Format (BCF) 文件的命令行工具集。它是与Samtools一起开发的,用于处理生物信息学中的DNA变异数据,例如单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)和插入/缺失变异(Insertions/Deletions,Indels)等。bcftools可以用于过滤、转换、合并、统计和分析这些变异数据。
以下是bcftools的一些常见用法:
- 格式转换: 使用bcftools可以将VCF文件转换为BCF文件或反向转换。这对于文件格式的处理和存储非常有用。
- 过滤变异: bcftools可以根据不同的标准过滤VCF/BCF文件中的变异。您可以根据质量分数、深度、基因型质量等参数来过滤变异。
- 合并文件: 如果您有多个VCF/BCF文件,bcftools可以帮助您将它们合并成一个文件,以进行后续的分析。
- 统计信息: 使用bcftools可以生成有关变异的统计信息,例如不同变异类型的计数、变异频率等。
- 基因型比较: 您可以使用bcftools比较不同样本之间的基因型,识别共享或不同的变异。
- 注释: 尽管bcftools本身不提供注释功能,但可以与其他工具(如VEP或Annovar)一起使用,以为变异添加注释信息。
- 区域选择: 您可以根据染色体位置或区域选择VCF/BCF文件中的特定变异。
- 过滤缺失值: 您可以使用bcftools过滤掉包含缺失基因型的变异。
- 计算AF值: AF(Allele Frequency)是指特定等位基因的频率。bcftools可以帮助您计算不同等位基因的频率。
bcftools的用法非常灵活,具体的命令和选项取决于您的分析需求。它通常通过命令行使用,您可以运行bcftools命令并附加子命令和选项来执行特定任务。建议查看bcftools的官方文档以获取详细的用法说明和示例。您可以在终端中输入bcftools --help来查看可用的子命令和选项列表。
bcftools的过滤变异的用法涉及到使用子命令bcftools filter,并提供适当的过滤条件。以下是详细的步骤和示例: - 基本语法:
bcftools filter [选项] 输入文件.vcf.gz -o 输出文件.vcf - 常见过滤条件:
- 质量过滤: 使用
-e选项来定义质量阈值,只保留质量得分高于阈值的变异。bcftools filter -e 'QUAL > 20' input.vcf.gz -o filtered.vcf - 深度过滤: 使用
-e选项来定义最小深度(DP)阈值,只保留深度大于阈值的变异。bcftools filter -e 'DP > 10' input.vcf.gz -o filtered.vcf - 基因型质量过滤: 使用
-e选项来定义最小基因型质量(GQ)阈值,只保留基因型质量高于阈值的变异。bcftools filter -e 'GQ > 30' input.vcf.gz -o filtered.vcf
- 质量过滤: 使用
- 多个过滤条件的组合: 您可以组合多个过滤条件来更精确地筛选变异。可以使用逻辑运算符(如
&&和||)来连接条件。bcftools filter -e 'QUAL > 20 && DP > 10' input.vcf.gz -o filtered.vcf - 排除变异: 如果您想排除符合条件的变异而不是保留它们,可以使用
-O选项来定义排除条件。bcftools filter -e 'QUAL < 10' -O t input.vcf.gz -o excluded.vcf - 特定区域的过滤: 如果您只想对特定染色体区域的变异进行过滤,可以使用
-r选项指定区域。bcftools filter -r chr1:1000-2000 -e 'QUAL > 20' input.vcf.gz -o filtered_region.vcf - 其他选项: bcftools还提供其他选项,例如基于频率的过滤和过滤特定基因型的变异。您可以查阅bcftools的官方文档以获取更多详细信息。
请根据您的具体需求修改上述示例中的过滤条件和选项。过滤条件应根据您的数据和研究问题进行调整,以确保您获得所需的变异集合。
很明显,是学徒把命令的规则弄错了:
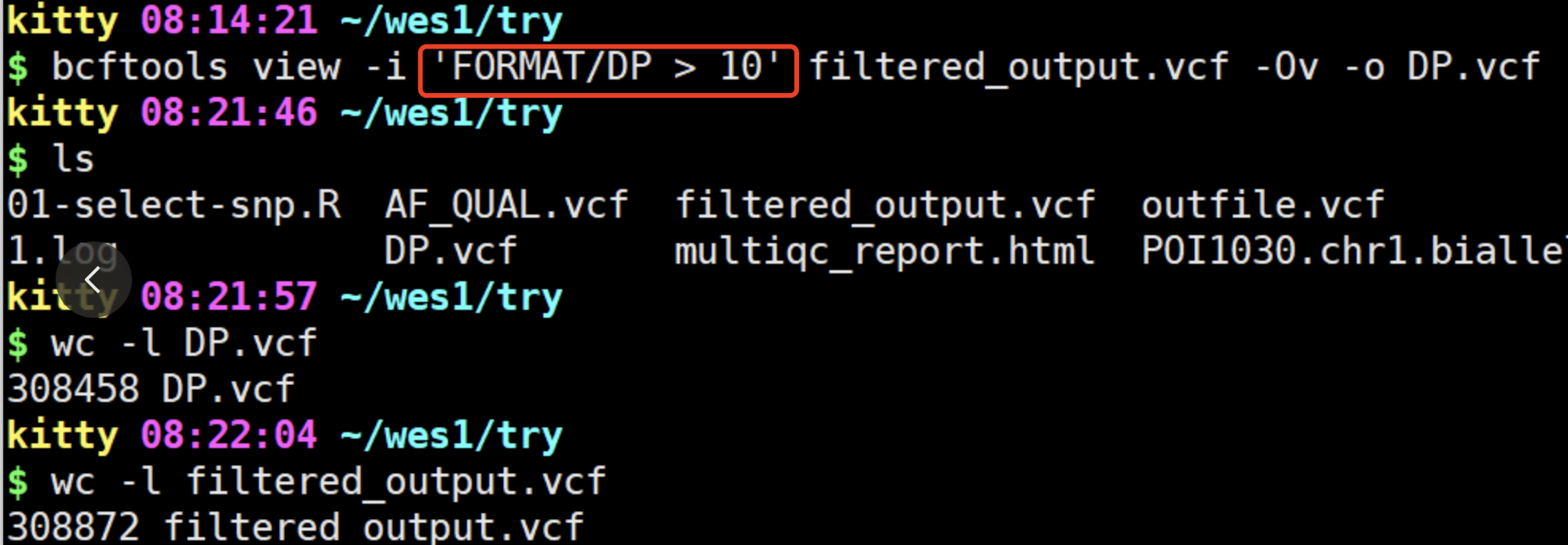
上面的错误的命令,很容易根据chatGPT给出来的回答,针对性的修改即可。