最近刷到了生物信息学领域著名期刊NAR的最新文章:《NORMSEQ – a tool for evaluation, selection and visualization of RNA-Seq normalization methods》是一个网页工具,NORMSEQ,它介绍了一些转录组测序表达量矩阵的归一化标准化方法学而且提供了一个在线网页工具给大家使用。
之所以注意到它,是因为NORMSEQ的流程图画的很吸引眼球:
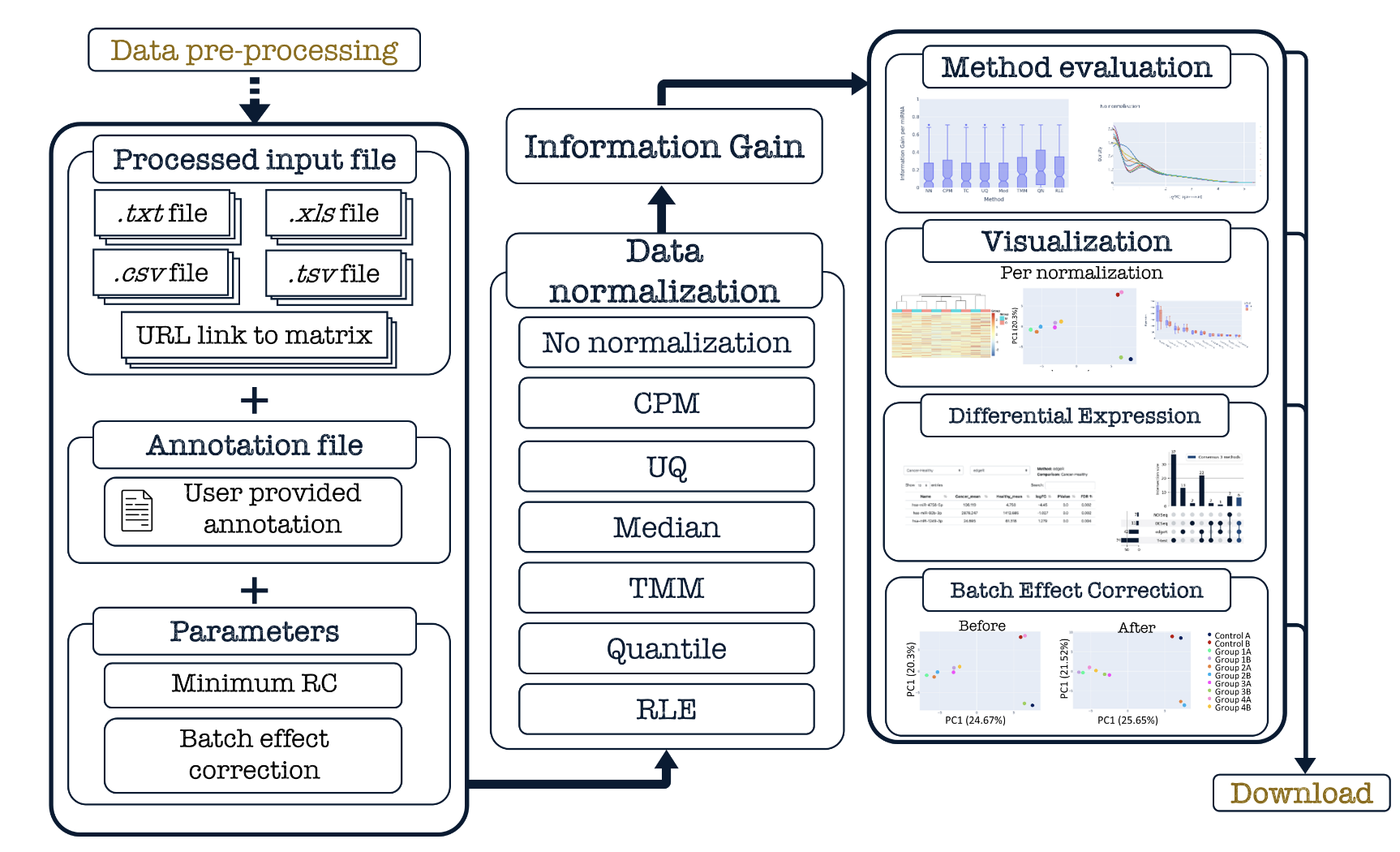
它起初是为了解决他们课题组自己的tRNA相关转录组测序表达量矩阵的归一化标准化问题,之前的文章是Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat Commun. 2020 Aug 14;11(1):4104. doi: 10.1038/s41467-020-17879-x. 对应的数据集是GSE141436. 是一个tRNA expression dataset ,里面有 21 different samples from seven mouse tissues ,这样的矩阵确实是超出了我的能力范围:
GSE141436_293FlpIn_DM_isodecoder_raw_counts.csv.gz 3.0 Kb
GSE141436_293FlpIn_isodecoder_raw_counts.csv.gz 12.1 Kb
GSE141436_Mouse_tissue_anticodon_raw_counts.csv.gz 3.6 Kb
GSE141436_Mouse_tissue_isodecoder_raw_counts.csv.gz 12.9 Kb
GSE141436_Mouse_tissue_variant_counts.csv.gz 720.5 Kb
GSE141436_Mouse_tissue_variant_counts_DM.csv.gz 136.2 Kb
现在它也可以针对普通的mRNA的转录组测序表达量矩阵的归一化标准化,提供如下所示:
- No normalization, just visualization
- Counts per Million
- Upper Quartile
- Median
- TMM
- Quantile
- Remove Unwanted Variation - RUVs
- Relative Log Expression
上面的每一个算法都对应着曾经“风靡一时”的普通的mRNA的转录组测序表达量矩阵的归一化标准化:
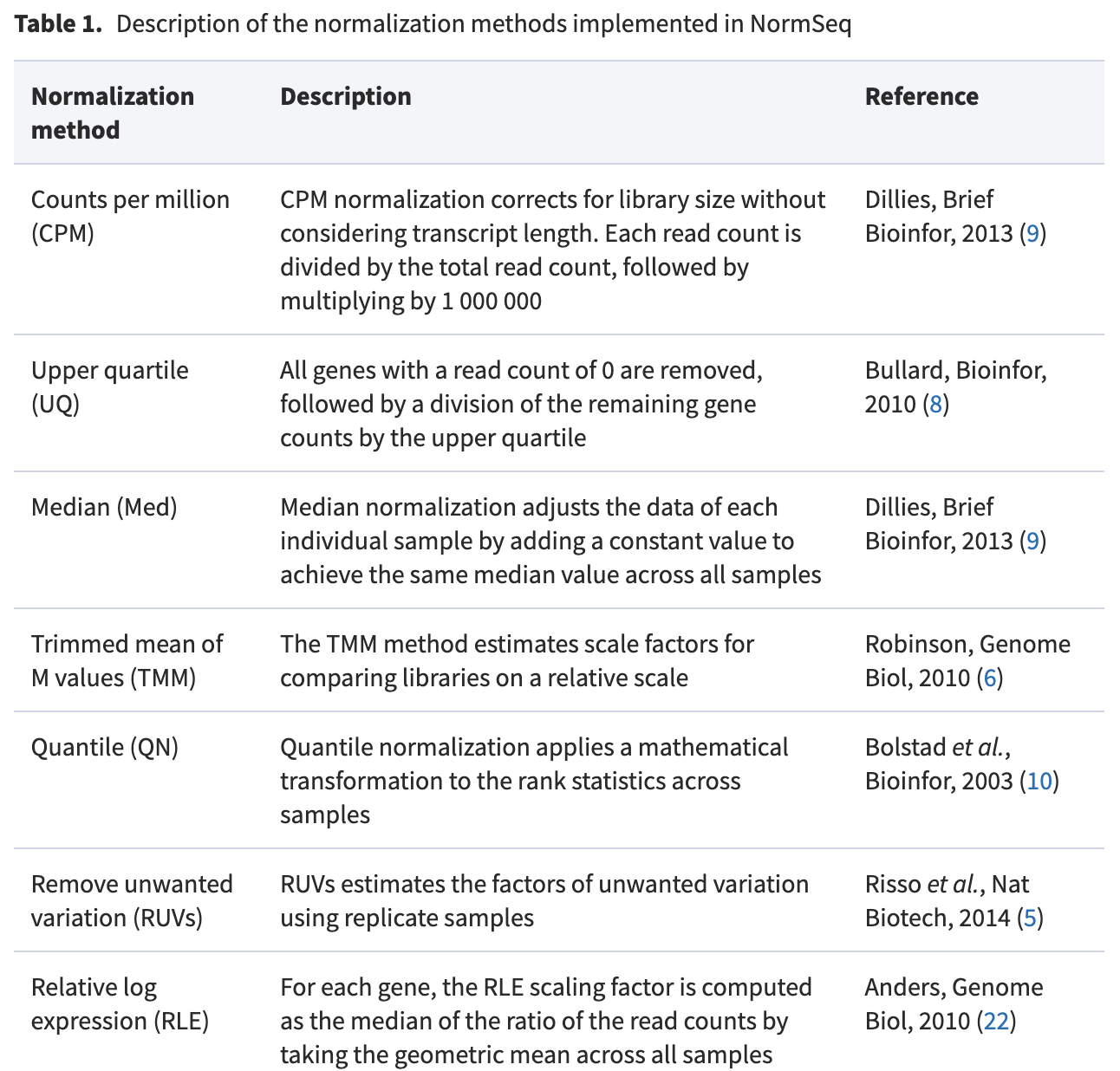
一般来说,我们简化这个普通的mRNA的转录组测序表达量矩阵的归一化标准化,我们最常用的是普通的CPM,就是,比如可以拿它来绘制PCA图:
colnames(symbol_matrix)
symbol_matrix[1:4,1:4] ## 基因名字的样品,矩阵
dat = log2(edgeR::cpm(symbol_matrix)+1)
dat[1:4,1:4]
boxplot(dat,las=2)
table(group_list)
exp=t(dat)#画PCA图时要求是行名时样本名,列名时探针名,因此此时需要转换
exp=as.data.frame(exp)#将matrix转换为data.frame
library("FactoMineR")#画主成分分析图需要加载这两个包
library("factoextra")
#~~~主成分分析图p2~~~
dat.pca <- PCA(exp , graph = FALSE)
this_title <- paste0(pro,'_PCA')
p2 <- fviz_pca_ind(dat.pca,
geom.ind = "point", #c( "point", "text" ), # show points only (nbut not "text")
col.ind = group_list, # color by groups
palette = "Dark2",
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups")+
ggtitle(this_title)+
theme(plot.title = element_text(size=12,hjust = 0.5))
实际上也可以首先把它这个普通的mRNA的转录组测序表达量矩阵的归一化标准化,不仅仅是普通的CPM矩阵形式:
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
library(data.table)
load(file = 'symbol_matrix.Rdata')
symbol_matrix[1:4,1:4]
library(limma)
library(edgeR)
library(DESeq2)
library(ggplot2)
load(file = 'symbol_matrix.Rdata')
symbol_matrix[1:4,1:4]
exprSet = symbol_matrix
(colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list) )
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
dds <- estimateSizeFactors(dds)
rld <- rlog(dds, blind=FALSE)
levels(rld$group_list)
assay(rld)[1:4,1:4]
#画PCA图时要求是行名时样本名,列名时探针名,因此此时需要转换
#将matrix转换为data.frame
exp=as.data.frame(t(assay(rld)))
exp[1:4,1:4]
library("FactoMineR")#画主成分分析图需要加载这两个包
library("factoextra")
#~~~主成分分析图p2~~~
dat.pca <- PCA(exp , graph = FALSE)
p2 <- fviz_pca_ind(dat.pca,
geom.ind = "point", #c( "point", "text" ), # show points only (nbut not "text")
col.ind = group_list, # color by groups
palette = "Dark2",
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups")+
theme(plot.title = element_text(size=12,hjust = 0.5))
但是从PCA的角度来看,基本上没什么区别,其实就是说前面列出来了的 Counts Per Million (CPM), Upper Quantile (UQ), Median (Med), Trimmed mean of M values (TMM), Quantile (QN), Relative Log Expression (RLE) and Remove Unwanted Variation in its RUVs version ,普通用户其实无需考虑那么多,任选其一即可,比如我就喜欢CPM格式进行后续可视化。
不过,仍然是需要值得注意的可能是 批次效应,如果有的话!!!
另外,其实我们并不喜欢在线网页工具,需要 uploading a file in txt, csv, tsv or xls formats 然后等网页返回处理好的结果,对我们来说就是首先得把数据从R里面写出来,然后结果再读入R里面,浪费时间。感兴趣的可以读一下它的源代码:
- NormSeq is freely available for all users at: https://arn.ugr.es/normSeq/
- the source code is available at https://github.com/cris12gm/normSeq.
是Python开发的哦!