众所周知,单个单细胞的转录组,如果是目前流行的10x这样的单细胞转录组技术,具体到每个细胞层面我们拿到的表达量信息通常是500到2000个基因的范围,也就是说如果我们的参考基因组注释文件里面是2万或者6万基因会出现起码90%的基因是缺失的,这个现象在单细胞转录组领域称作是drop-out (大家可以自行统计一下pbmc3k这个示例数据的drop-out情况 )
这一现象在单细胞测序中是常见的,它可能有以下几个原因:
- 低表达水平: 一些基因在单个细胞中的表达水平可能非常低,低到在测序中难以检测到。这种情况下,这些基因在某些细胞中会出现 “drop-out”。
- 技术限制: 单细胞RNA测序技术通常有其检测限制。某些基因可能由于技术上的限制而无法被准确地检测到。
- 细胞异质性: 单细胞RNA测序捕获的细胞是异质的,它们在基因表达方面存在差异。这意味着某些细胞可能表达某个基因,而其他细胞可能不表达,导致 “drop-out”。
- 样本处理: 样本的处理和准备也可能影响基因的检测。一些细胞可能在样本处理过程中失去或破坏RNA,从而导致某些基因的 “drop-out”。
但是,我们每个样品在10x技术层面可以出8000个左右的细胞,虽然具体的单个细胞表达的基因很少,但是作为一个整体,还是可以有2万多个基因的表达的,这就是为什么我们的表达量矩阵仍然是很大。
而我们的传统的转录组一般来说就是bulk层面,里面的细胞数量数不胜数,理论上不太可能出现大规模的0值,也就是说每个样品本来就是应该是有几万个基因有表达量的,这一点大家下载tcga或者gtex的表达量矩阵就可以验证一下!
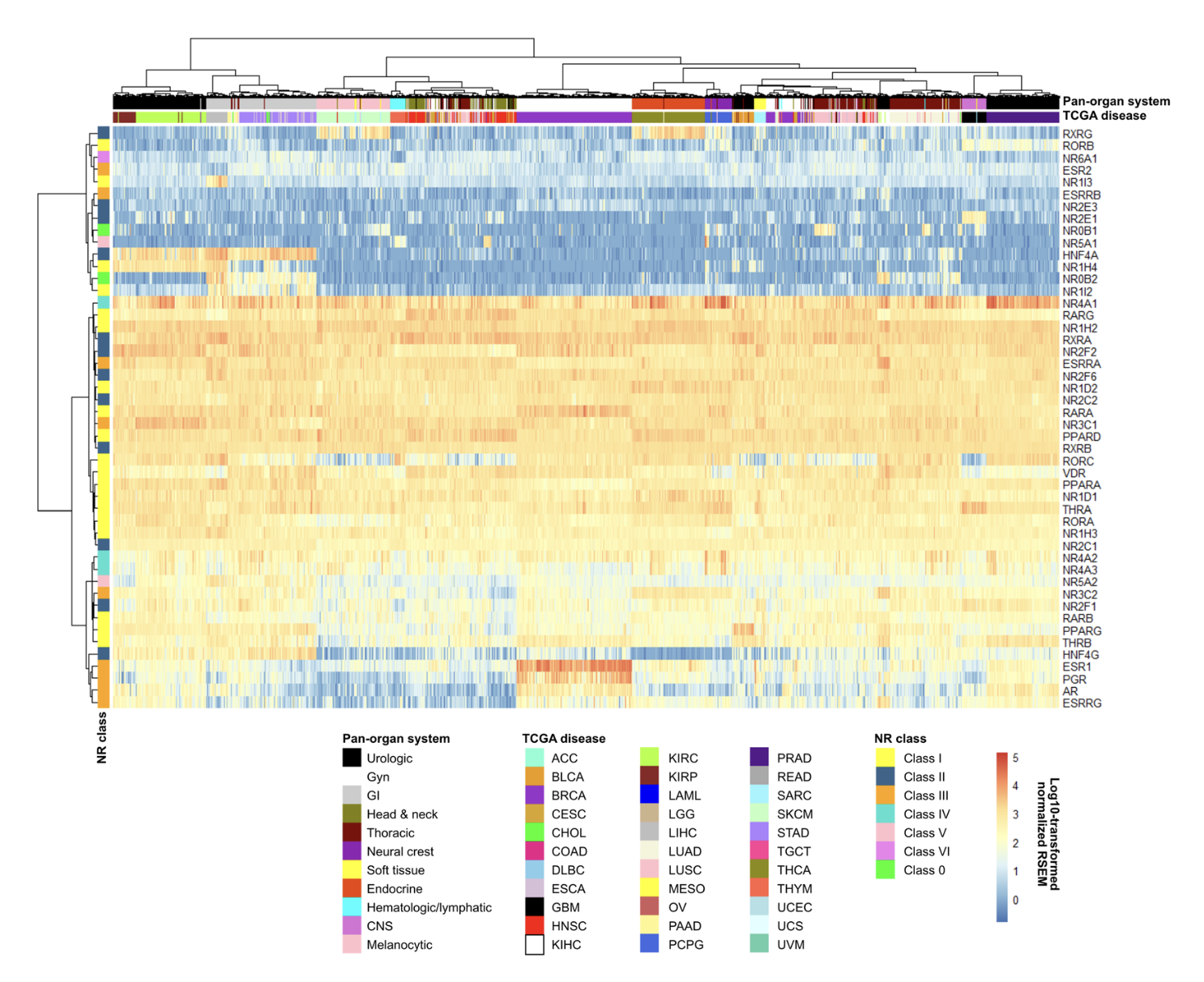
但是我看到了一个2020的泛癌数据挖掘文章:《pan-cancer analyses of human nuclear receptors reveal transcriptome diversity and prognostic value across cancer types》,作者就一个人, 他检查了 mRNA expression patterns for the 48 human NRs across 33 cancer types (8,526 TCGA patient samples )and 11 pan-organ groups
然后发现:
- 完全不表达的: five NRs (10%; ESR2, ESRRB, NR2E3, NR6A1, RORB) were not expressed in any tissue,
- 有癌症特异性的:22 NRs (46%; AR, ESR1, ESRRG, HNF4A, HNF4G, NR0B1, NR0B2, NR1H4, NR1I2, NR1I3, NR2E1, NR2F1, NR3C2, NR4A3, NR5A1, NR5A2, PGR, PPARG, RARB, RORC, RXRG, THRB)
- 在所有癌症都表达的:11 NRs (23%; NR1D1, NR1H3, NR2F2, NR2F6, NR3C1, NR4A2, PPARA, RARG, RORA, THRA, VDR) had widespread expression
- 在部分癌症表达的: 10 NRs (21%; ESRRA, NR1D2, NR1H2, NR2C1, NR2C2, NR4A1, PPARD, RARA, RXRA, RXRB) were ubiquitous.
很简单的一个热图就可以看到:
如果大家读了我们的系列教程:
- 有些批次效应是不可能被矫正的
- 不同癌症的差异难道大于其与正常对照差异吗
- 大样本量多分组表达量矩阵分析你难道没想到单细胞吗
- 泛癌分析时候的大样本量多分组建议选择tSNE而不是PCA
- 居然有如此多种癌症(是时候开启pan-cancer数据挖掘模式)
很容易做同样的分析看看,上面的数据挖掘文章居然得到了五个基因(ESR2, ESRRB, NR2E3, NR6A1, RORB)在所有的癌症样品都是不表达的,我怀疑里面应该是有错误。在所有样品都不表达的可能的原因有下面的这些:
- 低表达水平: 这些基因可能在样本中的表达水平非常低,低到标准测序技术无法准确检测到。在 bulk 转录组测序中,通常需要一定的表达量才能被检测到。
- 组织或细胞类型特异性: 某些基因可能仅在特定的组织或细胞类型中表达,而在其他组织或细胞中不表达。如果 bulk 转录组样本包括多种组织或细胞类型,那么这些基因在其中的表达可能会被稀释或掩盖。
- 技术限制: 某些标准转录组测序技术可能受到检测限制,无法准确检测低丰度的 mRNA。这可能导致一些基因的表达未被检测到。
- 数据处理或分析问题: 数据处理和分析过程中的错误或不正确的方法选择也可能导致未检测到某些基因的表达。这可能包括样本质量差、对数据进行过滤或修剪,或者使用了不适当的阈值。
- 基因缺失或突变: 某些基因可能由于突变或缺失而在样本中完全缺失。这些基因将不会在任何 bulk 转录组中表达。
但是那五个基因(ESR2, ESRRB, NR2E3, NR6A1, RORB)是 human nuclear receptors ,理论上不应该是有这样的情况发生。我简单查了一下,它确实是在 gencode.v22.annotation.gene.probeMap 文件里面,也就是说并不是因为基因被修改名字了,或者说也不是基因本身不在参考基因组注释文件造成的:
> b[match(cg,b$gene),]
id gene chrom chromStart chromEnd strand V2
ENSG00000140009.17 ESR2 chr14 64084232 64338112 - protein_coding
ENSG00000119715.13 ESRRB chr14 76310614 76501841 + protein_coding
ENSG00000278570.3 NR2E3 chr15 71792638 71818259 + protein_coding
ENSG00000148200.15 NR6A1 chr9 124517275 124771310 - protein_coding
ENSG00000198963.9 RORB chr9 74497365 74693177 + protein_coding
而且如果我们去看其它数据集里面的癌症样品,也会发现类似的现象,比如2022的文章《Clinical associations of ESR2 (estrogen receptor beta) expression across thousands of primary breast tumors》 就是重新发现了GSE96058数据集里面的ESR2基因几乎没有表达量。
The median expressi on wa s 0.05 log2(TPM + 0.1), and in 1027 samples ESR2 was not expressed at all.
现在已经是排除了数据处理或分析问题,然后普通转录组技术为什么会有这样的技术限制导致这5个基因被忽略呢?或者说它们本来就是超低水平基因?但是我们查询网页工具,发现它在正常组织其实是可以表达的(https://www.genecards.org/cgi-bin/carddisp.pl?gene=ESR2),所以就排除了组织或细胞类型特异性的原因,也不可能是转录组技术限制 :
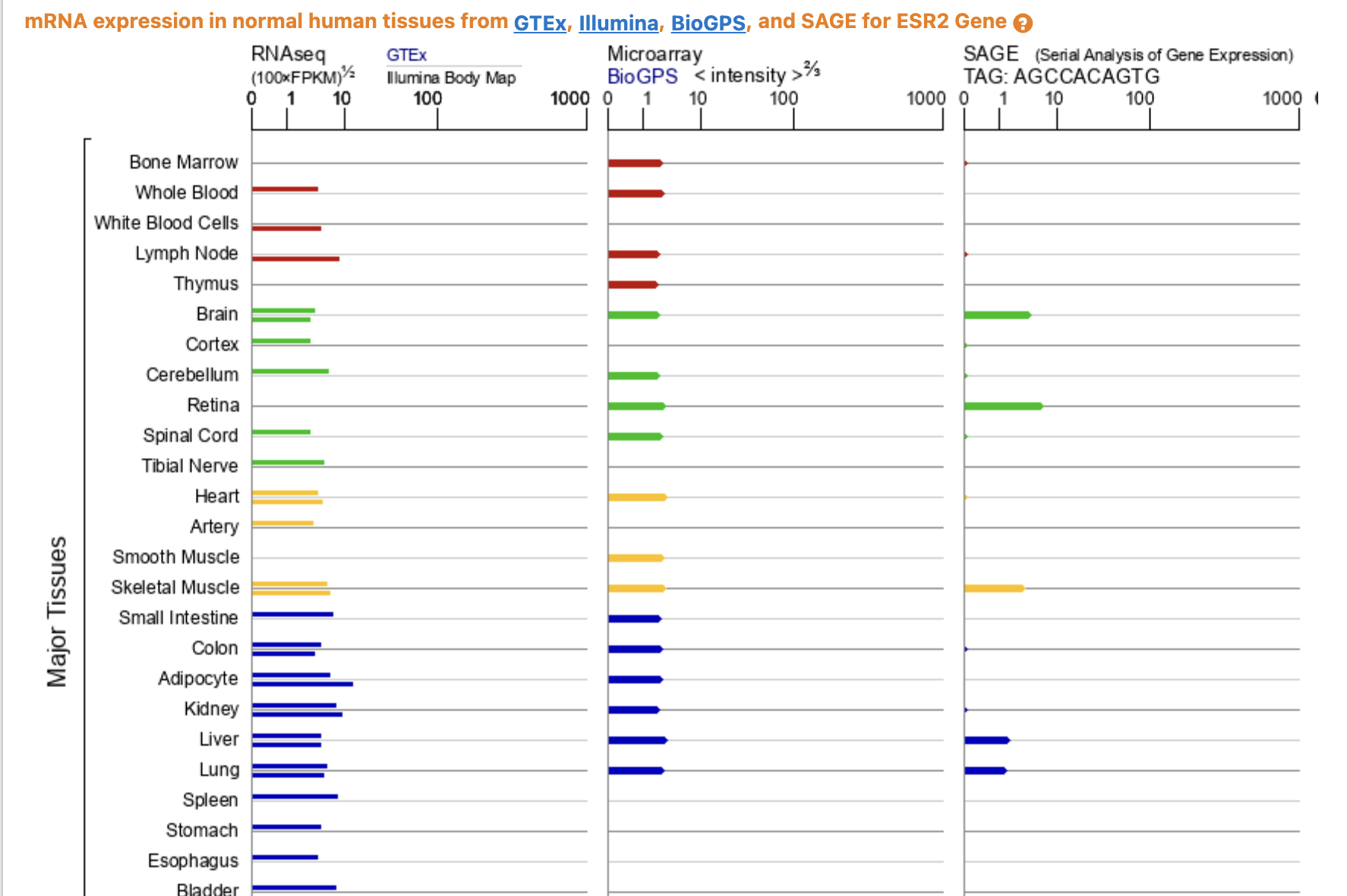
那么接下来还需要看它是不是恰好在所有的癌症都出现了拷贝数缺失,当然了这个概率实在是太小了。。。
不过柯南(福尔摩斯)曾经说过,当你排除一切不可能之后,剩下的那个无论多么令人难以置信……那就是真相!!!
类似的思路可以出现在任意的基因家族
人类有许多著名的基因家族,其中一些包括:
- Hox基因家族: Hox基因家族编码一组关键的转录因子,它们在胚胎发育中控制身体轴的分段和器官发育。这个家族在所有的脊椎动物中都是高度保守的。
- 免疫球蛋白家族: 免疫球蛋白家族包括抗体和T细胞受体等分子,它们在免疫系统中发挥着关键作用,用于识别和抵御病原体。
- 转录因子家族: 转录因子是一类控制基因表达的蛋白质,包括家族如bZIP、bHLH、homeodomain、和Zinc finger等。它们调控了许多生物学过程,包括发育、细胞增殖和分化。
- G蛋白偶联受体家族: G蛋白偶联受体家族是一组在细胞膜上的受体蛋白,它们参与了许多细胞信号传导通路,包括视觉、味觉和神经递质的感知。
- 核激素受体家族: 核激素受体家族包括雌激素受体、雄激素受体、甲状腺激素受体等,它们在细胞核中调控基因表达,对于生长、发育和代谢具有重要影响。
- Histone基因家族: 这个家族编码组蛋白,它们是染色质的主要组成部分,对基因的表达和染色质结构起关键作用。
- 细胞骨架蛋白家族: 这包括微管蛋白、中间丝蛋白和微丝蛋白等,它们在细胞的形态维持和运动中发挥重要作用。
这些基因家族在生物学研究中具有重要地位,因为它们控制着多种生物学过程,而它们的突变或异常表达通常与疾病有关。因此,对这些基因家族的研究对于理解生物学和开发治疗方法至关重要。
除了泛癌还可以在单细胞层面看基因家族的全部基因的情况
如果这些基因家族里面的基因在单细胞层面也有一些特性,就好玩了!