我首先让chatGPT帮我根据fasta格式的蛋白质序列文件,使用Python语言计算里面的每个蛋白质的序列长度和分子量,它给了描述了解题思路。。。

看起来蛮厉害的,我怀疑它可能是对我的真正的需求不够了解,我其实就是想让他帮忙写代码,就继续追问它:
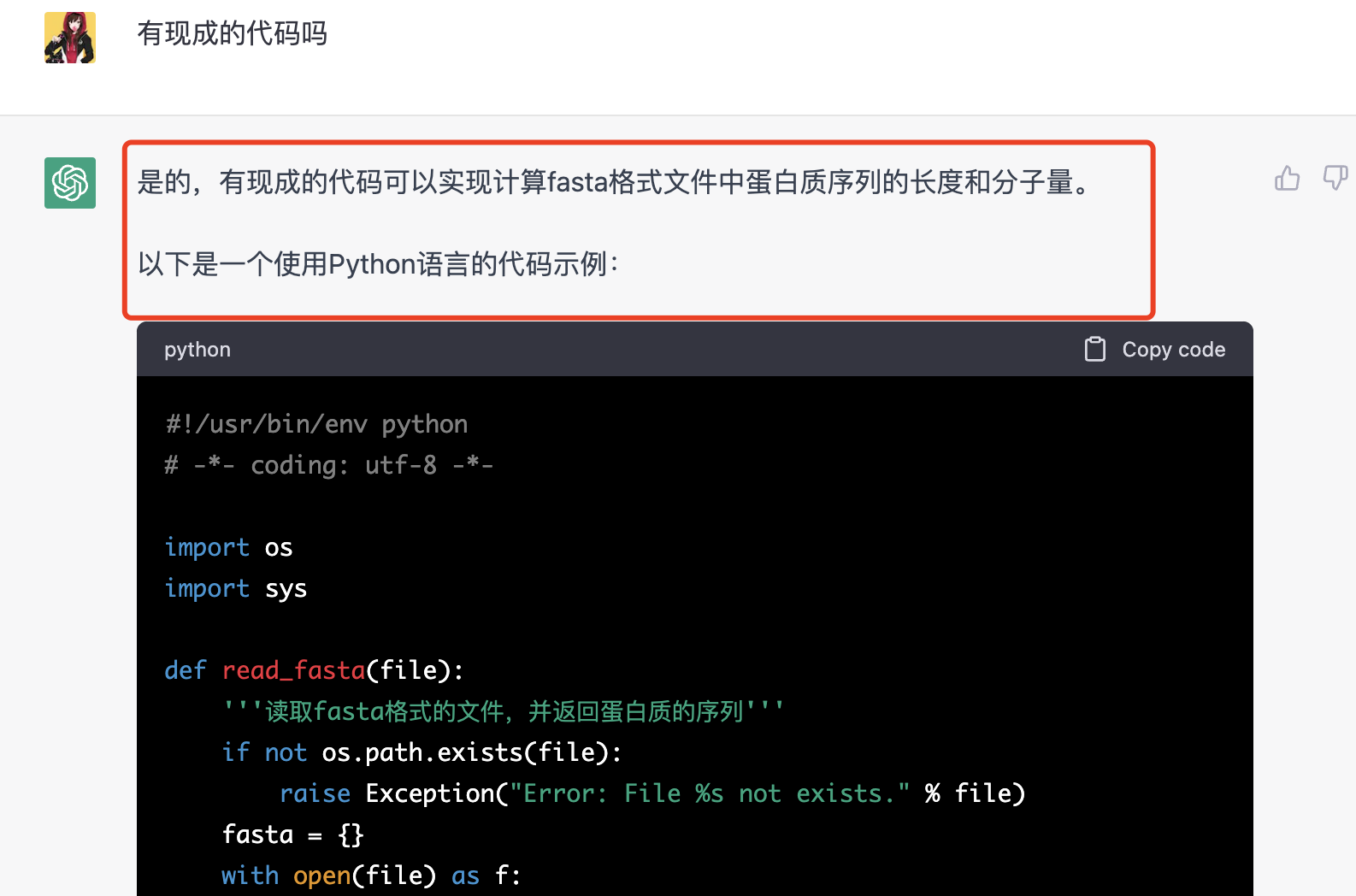
它居然创造了两个函数,Python能力已经超过了我,不知道它写的函数实用性如何,感兴趣的小伙伴可以复制粘贴去测试看看 :
#!/usr/bin/env python
# -*- coding: utf-8 -*-
import os
import sys
def read_fasta(file):
'''读取fasta格式的文件,并返回蛋白质的序列'''
if not os.path.exists(file):
raise Exception("Error: File %s not exists." % file)
fasta = {}
with open(file) as f:
name = ""
seq = ""
for line in f:
line = line.strip()
if line.startswith(">"):
if name:
fasta[name] = seq
name = line[1:]
seq = ""
else:
seq += line
fasta[name] = seq
return fasta
def get_molecular_weight(seq):
'''计算分子量'''
weight = 0
for aa in seq:
weight += aa_weight[aa]
return weight
aa_weight = {
'A': 89.09,
'R': 174.20,
'N': 132.12,
'D': 133.10,
'C': 121.15,
'E': 147.13,
'Q': 146.15,
'G': 75.07,
'H': 155.16,
'I': 131.17,
'L': 131.17,
'K': 146.19,
'M': 149.21,
'F': 165.19,
'P': 115.13,
'S': 105.09,
'T': 119.12,
'W': 204.23,
'Y': 181.19,
'V': 117.15
}
if __name__ == "__main__":
fasta = read_fasta(sys.argv[1])
for name, seq in fasta.items():
length = len(seq)
weight = get_molecular_weight(seq)
print("%s\t%d\t%.2f" % (name, length, weight))
是一个看起来很完整的Python脚本,使用Python解释器运行它就可以读取一个fasta格式的蛋白质序列文件然后做计算。(目前我不清楚这个代码是它在互联网搜索后拷贝给我的呢,还是它自己从零开始创造的, 如果是后者还是蛮让我震惊的。。。)
我简单的运行了一下, 找到了一个fasta格式的蛋白质序列文件测试了,前面的320个序列没有问题,中间会报错,不知道是不是我的文件格式不标准。。。
sp|Q96EX3|DC2I2_HUMAN Cytoplasmic dynein 2 intermediate chain 2 OS=Homo sapiens OX=9606 GN=DYNC2I2 PE=1 SV=2 536 67438.12
sp|Q9NS56|TOPRS_HUMAN E3 ubiquitin-protein ligase Topors OS=Homo sapiens OX=9606 GN=TOPORS PE=1 SV=1 1045 138004.09
Traceback (most recent call last):
File " test.py", line 61, in <module>
weight = get_molecular_weight(seq)
File " test.py", line 31, in get_molecular_weight
weight += aa_weight[aa]
KeyError: 'U'
当然了,如果完全没有编程经验的小伙伴就算是拿到了chatGPT帮忙写的如此优秀的代码,也不懂得如何运行,因为可能不知道在哪里运行Python命令。(而且, 遇到了报错也很难去解决。。。。)
其实肯定是有现成的轮子
没必要自己写两个函数,应该是有两个包可以完成这样的需求, 一个包可以读取fasta格式的蛋白质序列文件,一个包可以计算蛋白质序列的分子量,因为我对Python也不熟悉,所以就没有继续跟chatGPT对话,因为就算它给我们两个模块的名字我也很难判断它是不是在粗制滥造(瞎编乱造)。。。。
比如在R里面就有Biostrings和Peptides包,很容易做到:
#BiocManager::install('Biostrings')
library(Biostrings)
# 读取FASTA格式的蛋白质序列文件
sequences <- readAAStringSet("protein.fasta")
length(sequences)
# 计算每个蛋白质的序列长度
lengths <- sapply(sequences, nchar)
head(lengths)
# 计算每个蛋白质的分子量
# molecular_weights <- sapply(sequences, aa2mw)
# install.packages("Peptides", dependencies=TRUE)
library(Peptides)
mw(sequences[[1]])
lapply(head(sequences), mw)
其实,我到现在还觉得很震惊,对我这样的Python门外汉来说这么复杂的两个函数,它居然不费吹灰之力就写出来了。大家可以测试一下,其中 test.py 就是上面的Python代码,而test_protein.fasta大家很容易去找到啦;
python3 test.py test_protein.fasta
如果chatGPT继续进化下去,我们可能是最后一代大规模学编程的人呢,编程语言就跟之前的汇编语言一样,成为了历史,极少数人需要掌握。。。。
需要合理的描述需求
当然了,要熟练的使用chatGPT帮助我们来写代码做编程方面的工作,如何合理的提需求也是一个功夫活。我前面的需求其实很清晰, 就是用Python语言计算里面的每个蛋白质的序列长度和分子量。但是另外一个需求,比如:如何优雅的给单细胞转录组fastq文件改名,描述起来就很费劲,起码我觉得有难度,但是交流群小伙伴很快就给出来了提问方式:
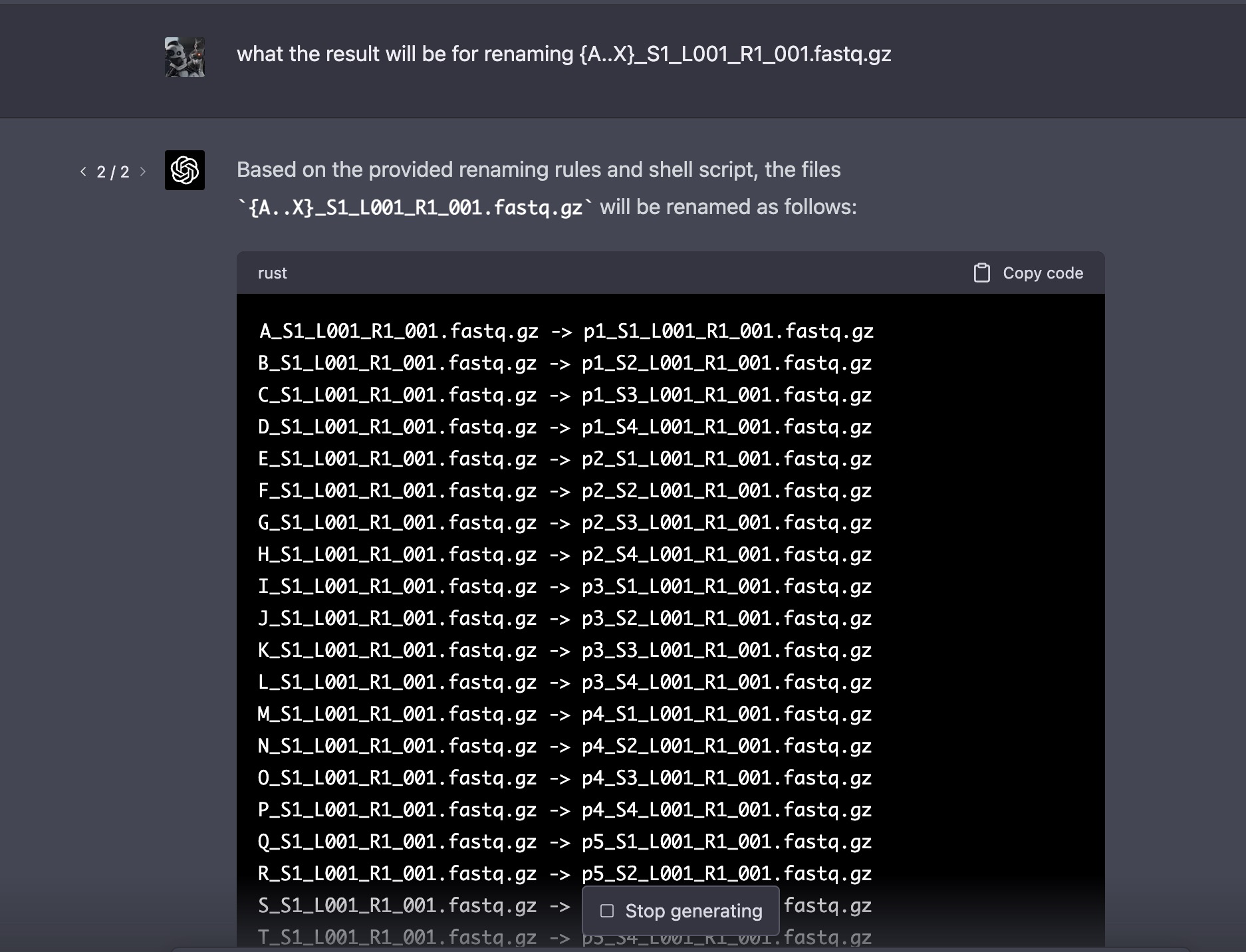
I have a series of files with names in the format {A..X}_S1_L001_R1_001.fastq.gz and {A..X}_S1_L001_R2_001.fastq.gz. I need to rename these files using the following rules:
Split the file name by underscores.
For the first part of the file name (y), replace it with px, where x is a number. Increment x by 1 after every 4 unique y values in alphabetical order, and reset the counter i to 0.
For the second part of the file name (Sz), replace it with Si, where i is a number. Increment i by 1 for each unique y value detected alphabetically.
chatGPT就真的顺利的给出来了一个非常复杂的shell脚本进行规则改名:
所以,大家到并不需要担心chatGPT会取代你们的工作,因为绝大部分老板根本就没办法说清楚自己的需求,压力仍然是要给到你们打工人,打工人就需要去跟chatGPT不停的沟通,不停地调试直到满足了老板的需求。如果老板可以直接说清楚需求,chatGPT就真正的抢走了你的饭碗。
