做生物信息学的我们经常会接触到生物信息学相关数据库等网络资源,绕不开的就是EBI和NCBI,我们以前介绍的单细胞转录组项目数据通常是ncbi旗下的sra或者geo数据库,但实际上ebi旗下的ArrayExpress数据库的也是有部分单细胞数据资源。
EBI (European Bioinformatics Institute) 和 NCBI (National Center for Biotechnology Information) 都是全球领先的生物信息学研究机构,它们提供了大量的生物信息学数据库和工具,对全球的科研工作者开放。
-
EBI 是欧洲生物信息学研究所的缩写,它是欧洲分子生物学实验室(EMBL)的一个分支机构。EBI 提供了许多重要的生物信息学资源,包括但不限于:Ensembl(基因注释),UniProt(蛋白质序列和功能信息),ArrayExpress(基因表达数据),PRIDE(蛋白质质谱数据),ChEMBL(化学和药物数据)等。
-
NCBI 是美国国立生物技术信息中心的缩写,它是美国国立卫生研究院(NIH)的一个部门。NCBI 提供了许多重要的生物信息学资源,包括但不限于:GenBank(核酸序列数据库),PubMed(生物医学文献数据库),BLAST(序列比对工具),dbSNP(单核苷酸多态性数据库),ClinVar(与人类变异相关的医学信息)等。
本次我们要处理的是ebi旗下的ArrayExpress数据库的E-MTAB-11450,其官方链接:https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-11450
有意思的是它也没有公开其单细胞转录组数据集的表达量矩阵,所以就需要从fastq文件开始,那就是可以看到: E-MTAB-11450.sdrf.txt 里面就有全部的样品的fq文件的ftp下载路径:
ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR870/ERR8701728/sc5rNAES015_hg19_S12_L001_I1_001.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR870/ERR8701728/sc5rNAES015_hg19_S12_L001_R1_001.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR870/ERR8701728/sc5rNAES015_hg19_S12_L001_R2_001.fastq.gz
只需要下载这些fastq文件的数据即可,然后走cellranger的定量流程。
cellranger的定量流程详解:
正常走cellranger的定量流程即可,代码我已经是多次分享了。参考:
- 10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元)
- 10X的单细胞转录组原始数据也可以在EBI下载
- 一个10x单细胞转录组项目从fastq到细胞亚群
- 一文打通单细胞上游:从软件部署到上游分析
- PRJNA713302这个10x单细胞fastq实战
- 一次曲折且昂贵的单细胞公共数据获取与上游处理
- 只能下载bam文件的10x单细胞转录组项目数据处理
- 不知道10x单细胞转录组样品和fastq文件的对应关系
- 10X单细胞转录组测序数据的 SRA转fastq踩坑那些事
- 10x的单细胞转录组fastq文件的R1和R2不能弄混哦
差不多几个小时就可以完成全部的样品的cellranger的定量流程。基础知识非常重要,我们在单细胞天地多次分享过cellranger流程的笔记(2019年5月),大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
首先需要在自己的服务器上面安装conda
自己下载安装自己的conda,自己配置哈。
# 首先下载文件,20M/S的话需要几秒钟即可
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
# 接下来使用bash命令来运行我们下载的文件,记得是一路yes下去
bash Miniconda3-latest-Linux-x86_64.sh
# 安装成功后需要更新系统环境变量文件
source ~/.bashrc
首先如果是在中国大陆,需要设置好镜像:
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
然后使用自己的conda来安装 kingfisher 软件:
conda install mamba
mamba create -n kingfisher python=3.8
mamba init
mamba list
# 重启terminal:
mamba activate kingfisher
mamba install -c bioconda kingfisher
前面是ftp地址,所以简单的wget或者curl命令即可,也可以使用axel等工具,值得注意的是下载数据的代码耗时很久,一般来说需要使用nohup包裹一下,或者说开启screen命令。大概会得到如下所示的文件:
ls -lh *z|cut -d" " -f 5-
231M 6月 9 22:36 sc5rNAES001_hg19_S16_L001_I1_001.fastq.gz
641M 6月 9 22:42 sc5rNAES001_hg19_S16_L001_R1_001.fastq.gz
1.6G 6月 9 22:50 sc5rNAES001_hg19_S16_L001_R2_001.fastq.gz
213M 6月 9 22:54 sc5rNAES002_hg19_S17_L001_I1_001.fastq.gz
593M 6月 9 22:59 sc5rNAES002_hg19_S17_L001_R1_001.fastq.gz
1.4G 6月 9 23:05 sc5rNAES002_hg19_S17_L001_R2_001.fastq.gz
511M 6月 9 23:10 sc5rNAES003_Hg19_S9_L001_I1_001.fastq.gz
1.6G 6月 9 23:16 sc5rNAES003_Hg19_S9_L001_R1_001.fastq.gz
3.8G 6月 9 23:26 sc5rNAES003_Hg19_S9_L001_R2_001.fastq.gz
397M 6月 9 23:31 sc5rNAES004_Hg19_S10_L001_I1_001.fastq.gz
1.1G 6月 9 23:37 sc5rNAES004_Hg19_S10_L001_R1_001.fastq.gz
2.7G 6月 9 23:48 sc5rNAES004_Hg19_S10_L001_R2_001.fastq.gz
但是参考 小鼠的5个样品的10x技术单细胞转录组上游定量(文末赠送全套代码),走过cellranger流程后,居然有4个样品失败了,如下所示:
==> log_sc5rNAES001_hg19.txt <== Exit status: 1
==> log_sc5rNAES002_hg19.txt <== Exit status: 1
==> log_sc5rNAES003_Hg19.txt <== Exit status: 0
==> log_sc5rNAES004_Hg19.txt <== Exit status: 0
==> log_sc5rNAES005_Hg19.txt <== Exit status: 0
==> log_sc5rNAES006_Hg19.txt <== Exit status: 0
==> log_sc5rNAES007_Hg19.txt <== Exit status: 0
==> log_sc5rNAES008_Hg19.txt <== Exit status: 0
==> log_sc5rNAES009_Hg19.txt <== Exit status: 0
==> log_sc5rNAES010_Hg19.txt <== Exit status: 0
==> log_sc5rNAES014_hg19.txt <== Exit status: 1
==> log_sc5rNAES015_hg19.txt <== Exit status: 1
状态为1的是失败了,也就是说前后各两个样品失败,我们打开其中一个失败的样品的日志文件看看:
[error] Pipestance failed. Error log at:
Log message:
Unable to distinguish between [SC5P-R2, SC3Pv2] chemistries based on the R2 read mapping for Sample sc5rNAES015_hg19
Total Reads = 100000
Mapped reads = 60503
Sense reads = 12082
Antisense reads = 19477
这个时候就需要debug了,我简单的问了一下chatGPT想试试看它能不能帮我debug,很明显这个chatGPT的回答仅仅是启发式的,还是得自己debug啊。。。。
这个错误信息表明,Cell Ranger无法根据R2读取的映射来区分10x Genomics的两种化学方法:SC5P-R2和SC3Pv2。这可能是由于输入数据的问题,或者是Cell Ranger版本的问题。
以下是一些可能的解决方案:
- 检查输入数据:确保你的输入数据是正确的,并且是使用10x Genomics的化学方法生成的。如果你的数据是由其他方法(如Drop-seq或inDrop)生成的,那么Cell Ranger可能无法正确处理。
- 指定化学版本:在运行Cell Ranger时,你可以使用
--chemistry选项来明确指定你的样本使用的化学版本。例如,如果你知道你的样本是使用SC5P-R2方法生成的,那么你可以在命令行中添加--chemistry=SC5P-R2。 - 更新Cell Ranger:如果你正在使用的Cell Ranger版本过旧,那么可能无法正确识别新的化学方法。你可以尝试更新到最新版本的Cell Ranger。
如果以上方法都不能解决问题,你可能需要联系10x Genomics的技术支持,他们可能能提供更具体的帮助。
所以,我们简单的看了看每个样品的测序数据fq文件的fastqc结果,首先是R1,仅仅是接头序列的,可以看到失败的4个样品它们的duplicate远高于成功的样品,这个可能是破局的关键信息。
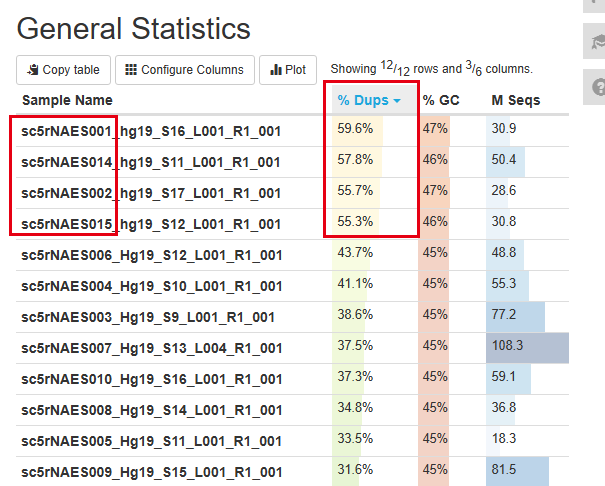
然后是R2,真正的测序片段数据文件:
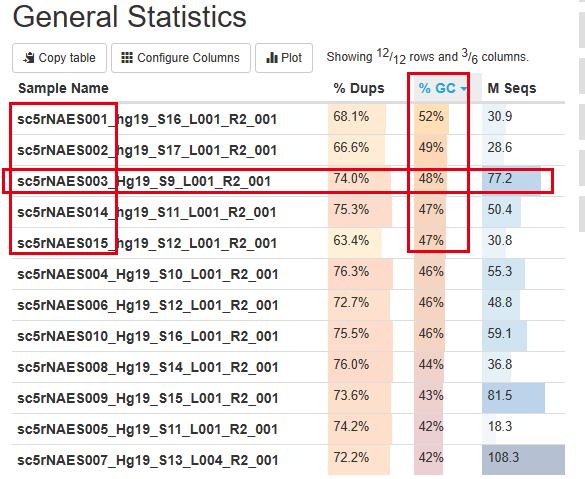
这个时候我们需要具体去检查每个样品的r1和r2,而不仅是看summary的表格:首先看其中一个失败的样品的r1的fastqc报表 :
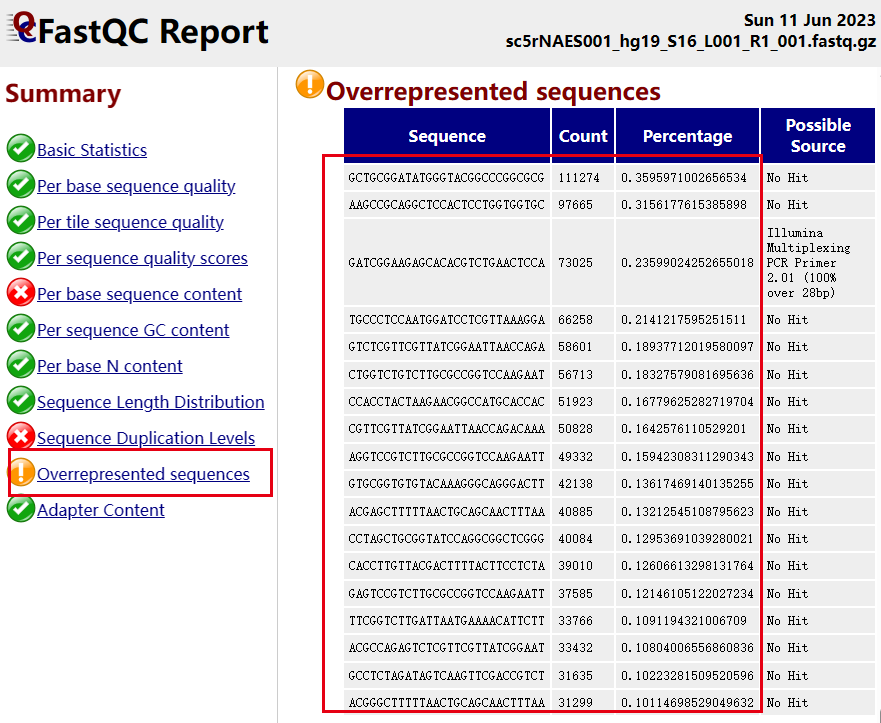
然后看其中一个成功的样品的r1的fastqc报表 ,去跟其中一个失败的样品的r1的fastqc报表可以对比。
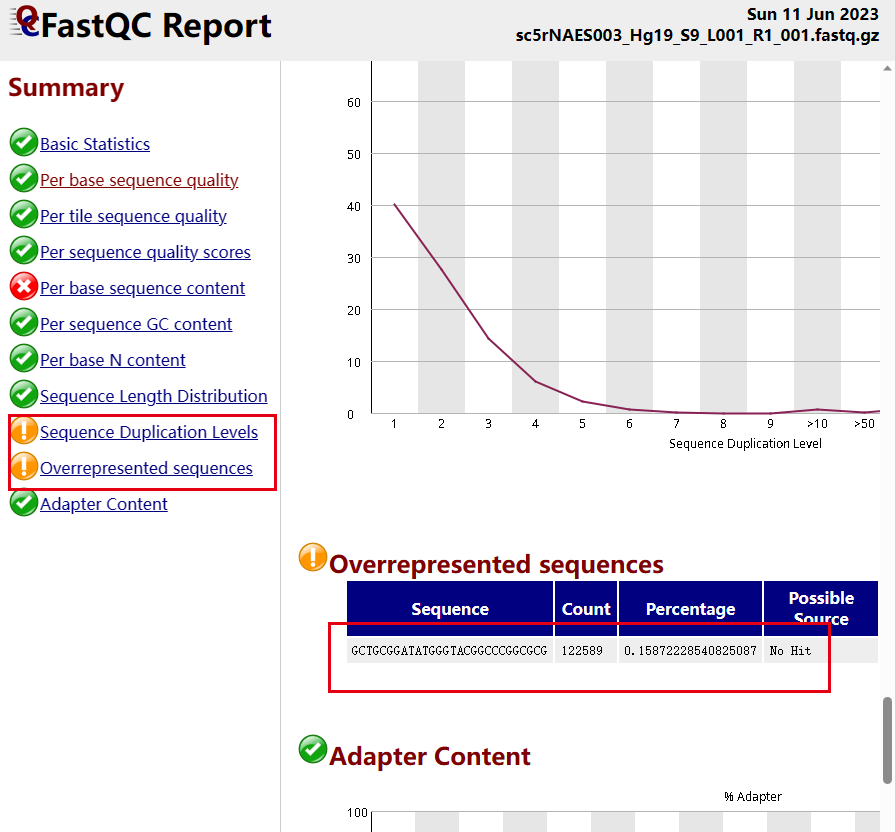
可以看到问题就出在R1文件,但是为什么失败的样品里面的r1如此奇怪呢?