在Seurat的官网可以看到SCTransform关于的描述,是:Note that this single command replaces NormalizeData(), ScaleData(), and FindVariableFeatures()
但是因为我接触单细胞有点早,是2017附近,那个时候经历了Seurat的v2变成v3的大更新,跟现在的小伙伴们经历了v4变成v5是一样的困扰,所以其实我从来就没有在我的代码里面做SCTransform,因为早期的 NormalizeData(), ScaleData(), FindVariableFeatures()三个函数,使用的也挺好的。但是最近学徒表示他发现了这里面的细节差异而且百思不得其解,所以我归纳汇总了一些学徒的探索,让大家一起看看是咋回事!
我们接下来使用的最简单的示范数据,来自于SeuratData包的ifnb数据集 :
rm(list = ls())
library(Seurat)
library(SeuratData)
# InstallData('ifnb.SeuratData')
# 使用上面的代码下载SeuratData包的ifnb数据集,但是非常考验网络。。。。
# trying URL 'http://seurat.nygenome.org/src/contrib/ifnb.SeuratData_3.1.0.tar.gz'
# Content type 'application/octet-stream' length 413266233 bytes (394.1 MB)
# 如果网络不行,就想办法下载(394.1 MB)保存在自己的电脑里面,然后下面的代码安装指定路径的压缩包:
# install.packages(pkgs = '~/database/seurat-data/ifnb.SeuratData_3.1.0.tar.gz',
# repos = NULL,
# type = "source" )
# 一定要看到这个包成功安装哦
# * DONE (ifnb.SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
table(ifnb$orig.ident)
table(ifnb$seurat_annotations,ifnb$orig.ident)
可以看到这个数据集是两个样品(两个处理)的单细胞转录组,而且是被提前注释完毕了的:
IMMUNE_CTRL IMMUNE_STIM
CD14 Mono 2215 2147
CD4 Naive T 978 1526
CD4 Memory T 859 903
CD16 Mono 507 537
B 407 571
CD8 T 352 462
T activated 300 333
NK 298 321
DC 258 214
B Activated 185 203
Mk 115 121
pDC 51 81
Eryth 23 32
接下来我们就使用不同的策略来走降维聚类分群流程哈!
首先看看SCTransform后的harmony整合前后效果
library(Seurat)
library(SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
sce <- SCTransform(ifnb,verbose = F)
sce <- RunPCA(sce,verbose = F)
sce <- RunUMAP(sce, dims = 1:20)
p1 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
library(harmony)
sce <- RunHarmony(sce, "orig.ident")
sce <- RunUMAP(sce, dims = 1:25,
reduction = "harmony")
p2 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
可以看到,这个时候我们选择了SCTransform函数,而且是可视化了harmony整合前后效果,如下所示:
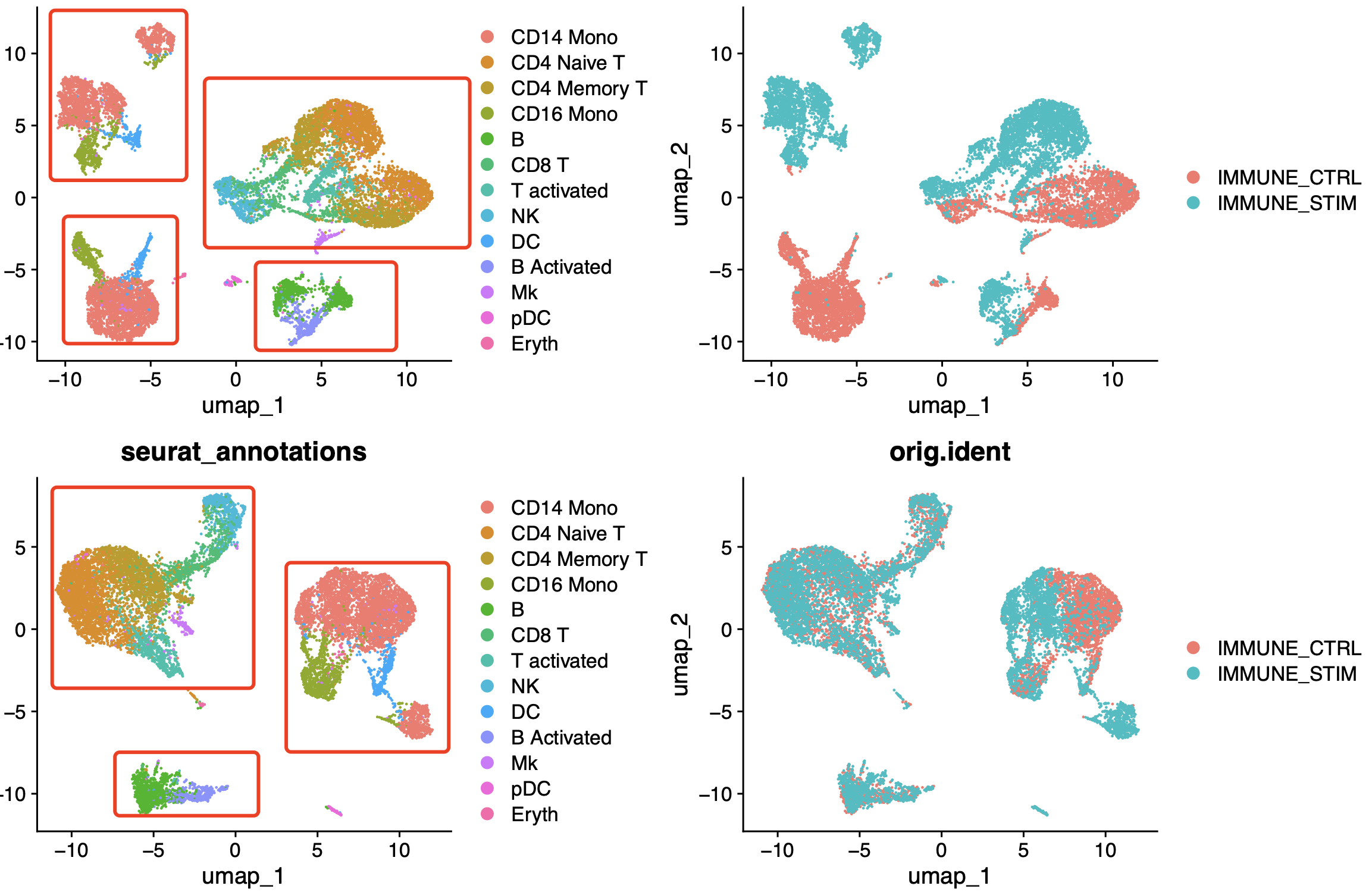
可以很清晰的看到,如果是不使用harmony整合就是上面的UMAP的二维可视化图,那么两个样品的t和b淋巴细胞其实是可以勉强融合在一起的,但是髓系淋巴细胞就差很远了。但是下面的UMAP的二维可视化图可以看到harmony整合后确实是髓系淋巴细胞可以跨越两个样品被融合在一起了。
接下来看看不使用SCTransform的对比
library(Seurat)
library(SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
input_sce <- NormalizeData(ifnb,
normalization.method = "LogNormalize",
scale.factor = 1e4)
input_sce <- FindVariableFeatures(input_sce)
input_sce <- ScaleData(input_sce)
input_sce <- RunPCA(input_sce, features = VariableFeatures(object = input_sce))
sce <- RunUMAP(input_sce, dims = 1:20)
p3 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
library(harmony)
sce <- RunHarmony(sce, "orig.ident")
sce <- RunUMAP(sce, dims = 1:25,
reduction = "harmony")
p4 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
p1/p2
ggplot2::ggsave('SCTransform-then-harmony-all-celltype.pdf',width = 12,height = 8)
p3/p4
ggplot2::ggsave('NormalizeData-then-harmony-all-celltype.pdf',width = 12,height = 8)
从上面的代码可以看到这个时候我们仍然是使用了早期的 NormalizeData(), ScaleData(), FindVariableFeatures()三个函数,所以harmony整合与否的UMAP的二维可视化图如下所示:

同样的,在harmony之前两个样品的 髓系淋巴细胞有样品特异性,但是harmony整合后确实是髓系淋巴细胞可以跨越两个样品被融合在一起了。而且从右图我们可以看到,其实是比使用SCTransform的流程的对两个样品的整合要好一点!
如果仅仅是针对单核细胞走流程呢
因为很多时候我们会提取自己的单细胞转录组数据里面的每个亚群做同样的分析,也会发现不同的参数不同的函数,效果是千差万别。我们也是测试一下:
rm(list = ls())
library(Seurat)
library(SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
kp = ifnb$seurat_annotations %in% c('CD14 Mono' , 'CD16 Mono')
ifnb=ifnb[,kp]
sce <- SCTransform(ifnb,verbose = F)
sce <- RunPCA(sce,verbose = F)
sce <- RunUMAP(sce, dims = 1:20)
p1 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
library(harmony)
sce <- RunHarmony(sce, "orig.ident")
sce <- RunUMAP(sce, dims = 1:25,
reduction = "harmony")
p2 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
library(Seurat)
library(SeuratData)
library(ifnb.SeuratData)
data("ifnb")
ifnb=UpdateSeuratObject(ifnb)
kp = ifnb$seurat_annotations %in% c('CD14 Mono' , 'CD16 Mono')
ifnb=ifnb[,kp]
input_sce <- NormalizeData(ifnb,
normalization.method = "LogNormalize",
scale.factor = 1e4)
input_sce <- FindVariableFeatures(input_sce)
input_sce <- ScaleData(input_sce)
input_sce <- RunPCA(input_sce, features = VariableFeatures(object = input_sce))
sce <- RunUMAP(input_sce, dims = 1:20)
p3 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
library(harmony)
sce <- RunHarmony(sce, "orig.ident")
sce <- RunUMAP(sce, dims = 1:25,
reduction = "harmony")
p4 = DimPlot(sce,group.by = 'seurat_annotations') +
DimPlot(sce,group.by = 'orig.ident')
p1/p2
ggplot2::ggsave('SCTransform-then-harmony-mono.pdf',width = 12,height = 8)
p3/p4
ggplot2::ggsave('NormalizeData-then-harmony-mono.pdf',width = 12,height = 8)
可以看到SCTransform函数配合harmony效果如下所示:
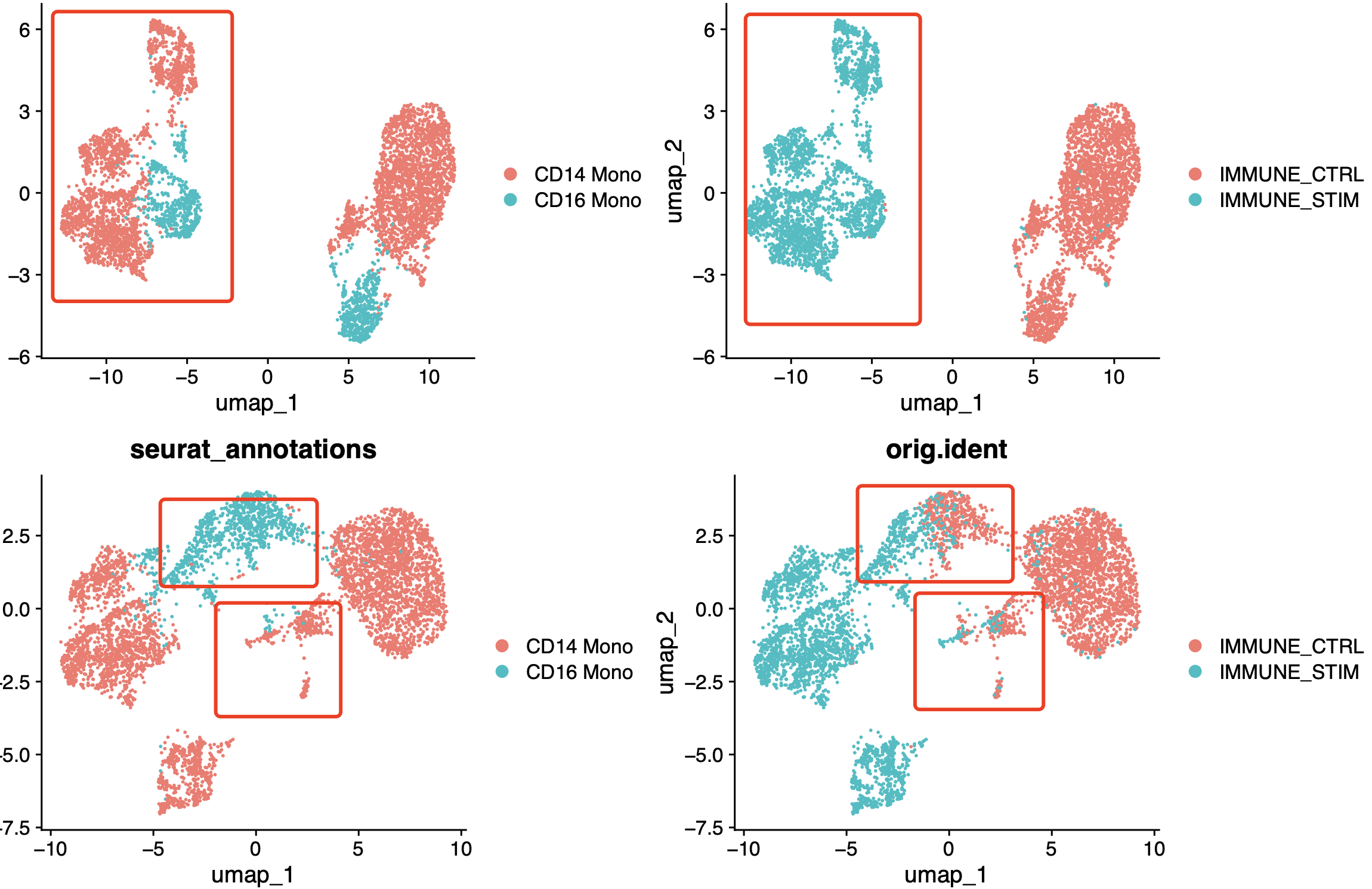
也就是说,两个样品的髓系淋巴细胞有样品特异性,但是我们的harmony整合可以让两个样品cd16单核在UMAP的二维可视化图靠近,以及两个样品的一小撮cd14单核在UMAP的二维可视化图靠近,但是很明显,STIM组里面的cd14单核还是有3个顽强的独立的亚群,是它比较特有的。
让我们看看传统的 NormalizeData(), ScaleData(), FindVariableFeatures()三个函数的效果吧!
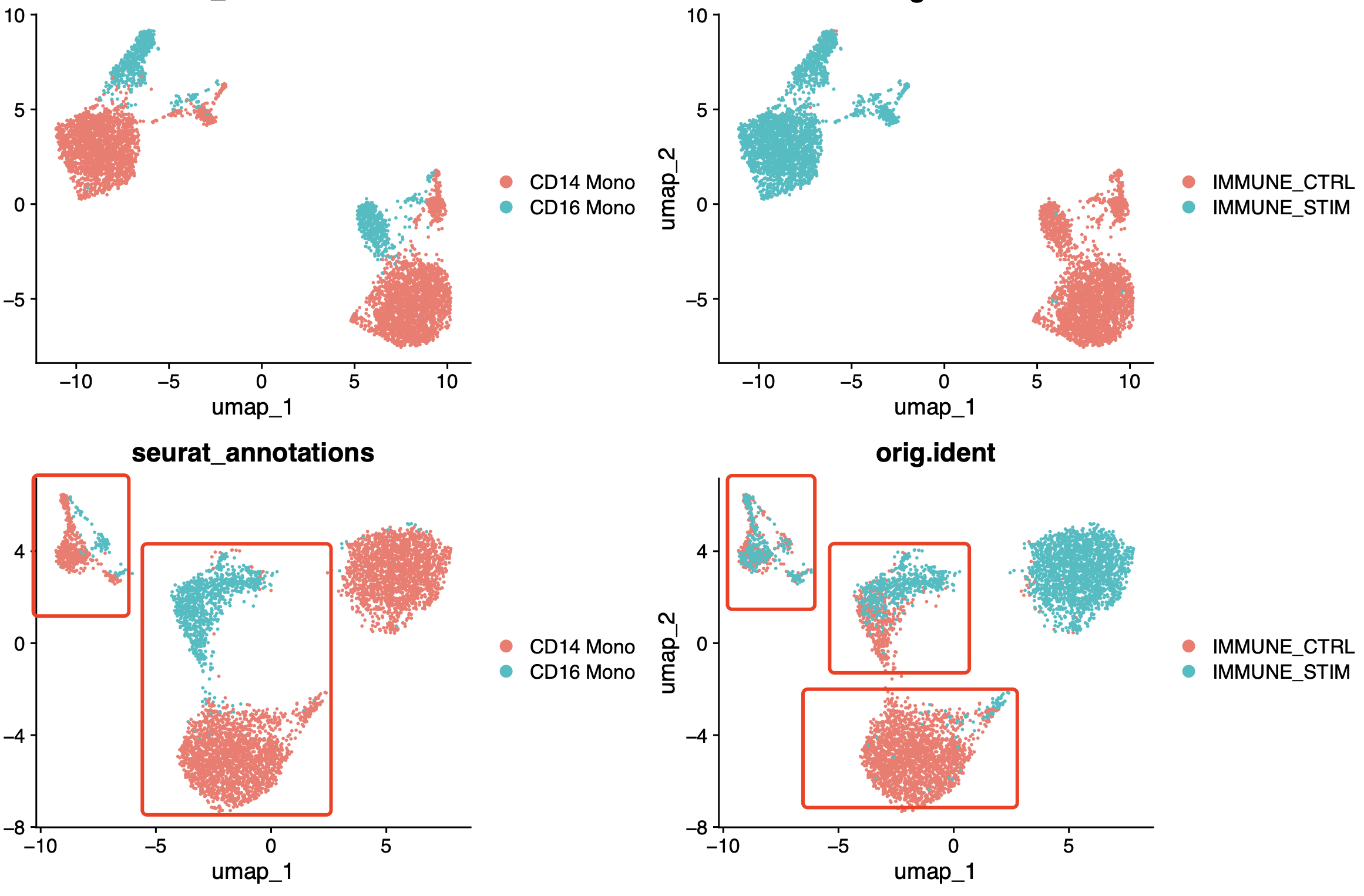
同样的,如果不使用harmony那么肯定是样品的异质性体现的淋漓尽致,但是我们的harmony整合在这个时候似乎是比上面的CTransform函数配合harmony效果要好一点点!
也可以换一个角度去对比
比如同样的是harmony整合, 但是如果是针对全部的单细胞亚群组合的数据集,可以看到来源于两个样品的CD14和CD16的单核细胞混合的非常好。但是如果是提取了单核细胞子集后,就可以很明显的看到cd14单核细胞的异质性了!
那么这个cd14单核细胞的异质性是否有生物学解释呢?
rm(list=ls())
options(stringsAsFactors = F)
source('scRNA_scripts/lib.R')
###### step1:导入数据 ######
sce.all.int = readRDS('../2-harmony/sce.all_int.rds')
colnames(sce.all.int@meta.data)
table(sce.all.int$seurat_annotations)
sce.all=sce.all.int[,sce.all.int$seurat_annotations %in% c('CD14 Mono' , 'CD16 Mono') ]
sce.all=CreateSeuratObject(
counts = sce.all@assays$RNA@counts,
meta.data = sce.all@meta.data
)
###### step2:QC质控 ######
dir.create("./1-QC")
setwd("./1-QC")
# 如果过滤的太狠,就需要去修改这个过滤代码
source('../scRNA_scripts/qc.R')
sce.all.filt = basic_qc(sce.all)
print(dim(sce.all))
print(dim(sce.all.filt))
setwd('../')
sp='human'
###### step3: harmony整合多个单细胞样品 ######
if(T){
dir.create("2-harmony")
getwd()
setwd("2-harmony")
source('../scRNA_scripts/harmony.R')
# 默认 ScaleData 没有添加"nCount_RNA", "nFeature_RNA"
# 默认的
sce.all.int = run_harmony(sce.all.filt)
setwd('../')
}
###### step4: 看标记基因库 ######
# 原则上分辨率是需要自己肉眼判断,取决于个人经验
dir.create('check-by-0.1')
setwd('check-by-0.1')
sel.clust = "RNA_snn_res.0.1"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
我们使用什么的标准的降维聚类分群代码试试看,可以得到如下所示:
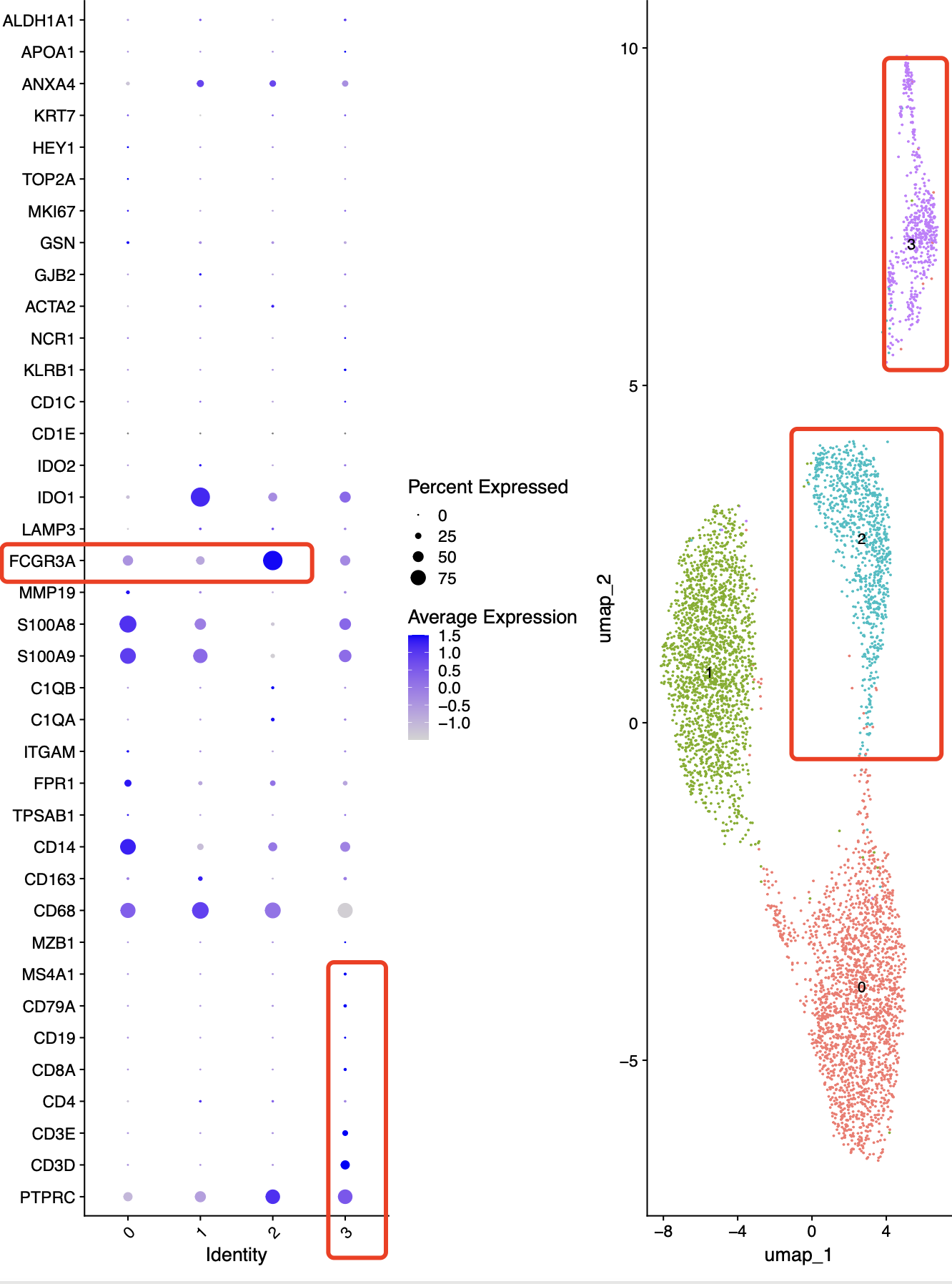
可以看到cd16确实是一个比较纯粹的单核细胞亚群,但是cd14就是很明显的两个亚群,那么我们看看它是否有什么区别呢?如下所示,可以看到的是1这个亚群其实是比较特殊的cd14单核细胞,因为它干扰素相关基因表达量过高,而且质量要差一点!而3这个亚群,它其实是被t和b这样的淋巴细胞给影响了而不再是一个纯粹的髓系免疫细胞啦!

而且我们可以看看这些单细胞亚群的样品分布情况:
IMMUNE_CTRL IMMUNE_STIM
0 1987 70 (cd14单核细胞亚群)
1 26 1866( cd14单核细胞亚群)
2 431 480 (cd16单核细胞亚群)
3 274 265 (淋巴细胞污染)
可以看到的是cd16单核细胞亚群确实是被harmony整合的挺好的,在UMAP的二维可视化图两个样品来源的cd16单核细胞靠很近。而cd14单核细胞,它在两个样品里面截然不同,哪怕是harmony也无法整合起来,也就是说STIM组相对于CTRL组来说,它里面cd14单核细胞已经是发生了生物学本质的变化!但是如果你仔细看上面的热图,其实cd16里面的也是有大量的干扰素相关基因表达量过高,只不过是在大环境层面的降维聚类分群因为cd16单核数量远少于cd14,所以它异质性没那么强到需要分成两个亚群,或者说我们的分辨率不够高。而且从cd14的角度来看,编号为1的亚群甚至都有点勉强了,而且STIM组相对于CTRL组来说cd14信号也未免太强了。。。。
但是cd16就非常统一了,哪怕是同样的受到了STIM组大量的干扰素相关基因表达量过高的影响:
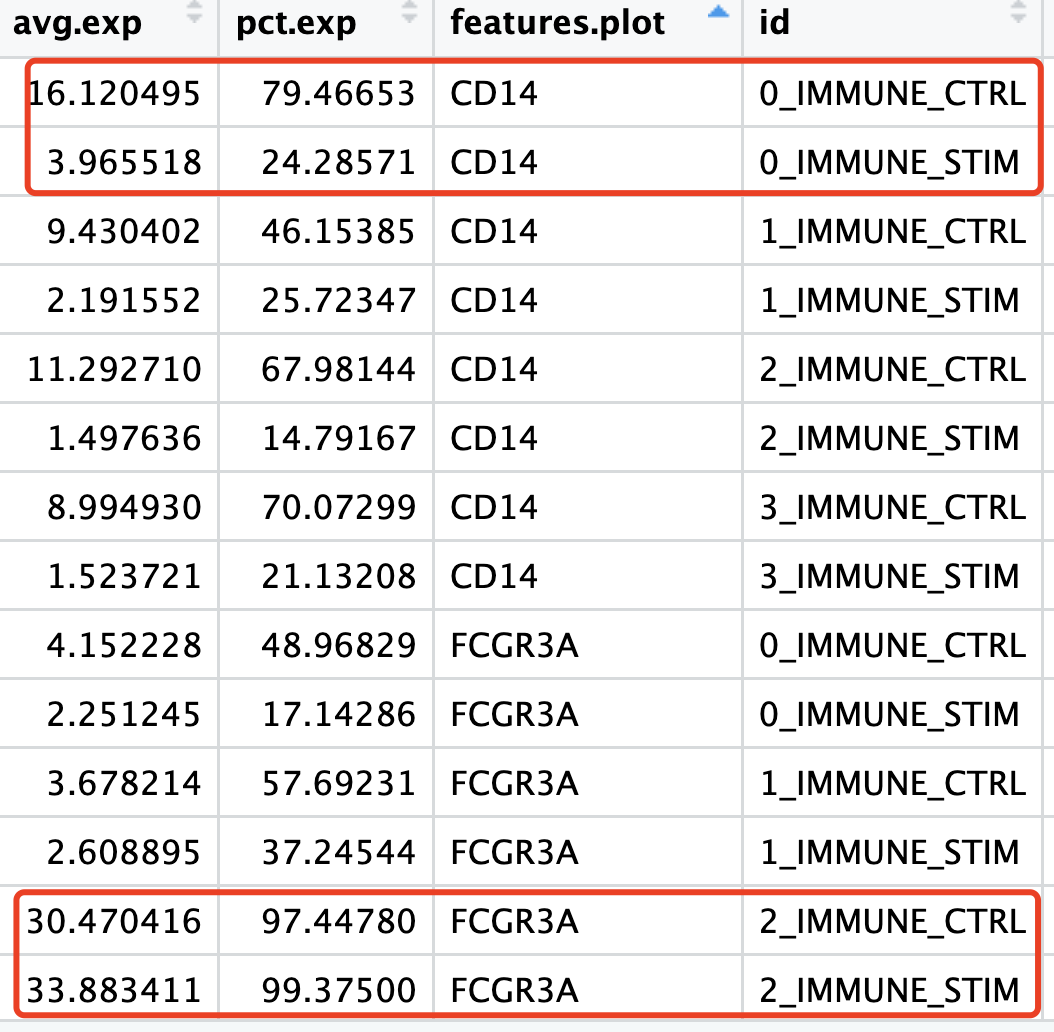
小结一下
第一层次降维聚类分群的时候,做SCTransform确实是可以代替早期的 NormalizeData(), ScaleData(), FindVariableFeatures()三个函数,但是在具体的单细胞亚群细分的时候,个人觉得SCTransform表现并不好,可能是因为它是针对完整的单细胞表达量矩阵设计的?或者说应该是在具体的每个样品内部跑SCTransform后然后再多个样品合并?