KEGG数据库是一个综合性的生物信息数据库,由日本京都大学生物信息学中心的Kanehisa实验室于1995年建立。它整合了基因组、化学和系统功能信息,旨在从分子水平上理解生物系统的高级功能和实用程序,特别是细胞、生物体和生态系统的功能。
KEGG数据库的分类情况相当广泛,它将信息分为三大类:系统信息、基因组信息和化学信息。进一步细分为16个主要的数据库。这些数据库包括:
- 系统信息:涉及生物系统的高级功能和实用程序,如KEGG PATHWAY、KEGG BRITE、KEGG MODULE、KEGG DISEASE、KEGG DRUG和KEGG ENVIRON等。
- 基因组信息:包括KEGG ORTHOLOGY、KEGG GENOME、KEGG GENES和KEGG SSDB等,这些数据库提供了基因组和基因相关的信息。
- 化学信息:如KEGG COMPOUND、KEGG GLYCAN、KEGG REACTION等,这些数据库包含了关于化学物质、代谢物、生化反应等的信息。
虽然KEGG数据库看起来非常复杂,但是绝大部分让接触到KEGG数据库其实是里面的 PATHWAY数据库。
什么是KEGG PATHWAY数据库
这个是是一个手工绘制的代谢通路集合,它包含了分子间相互作用和反应网络的信息,覆盖了新陈代谢、环境信息加工、细胞过程、生物体系统和人类疾病等方面。
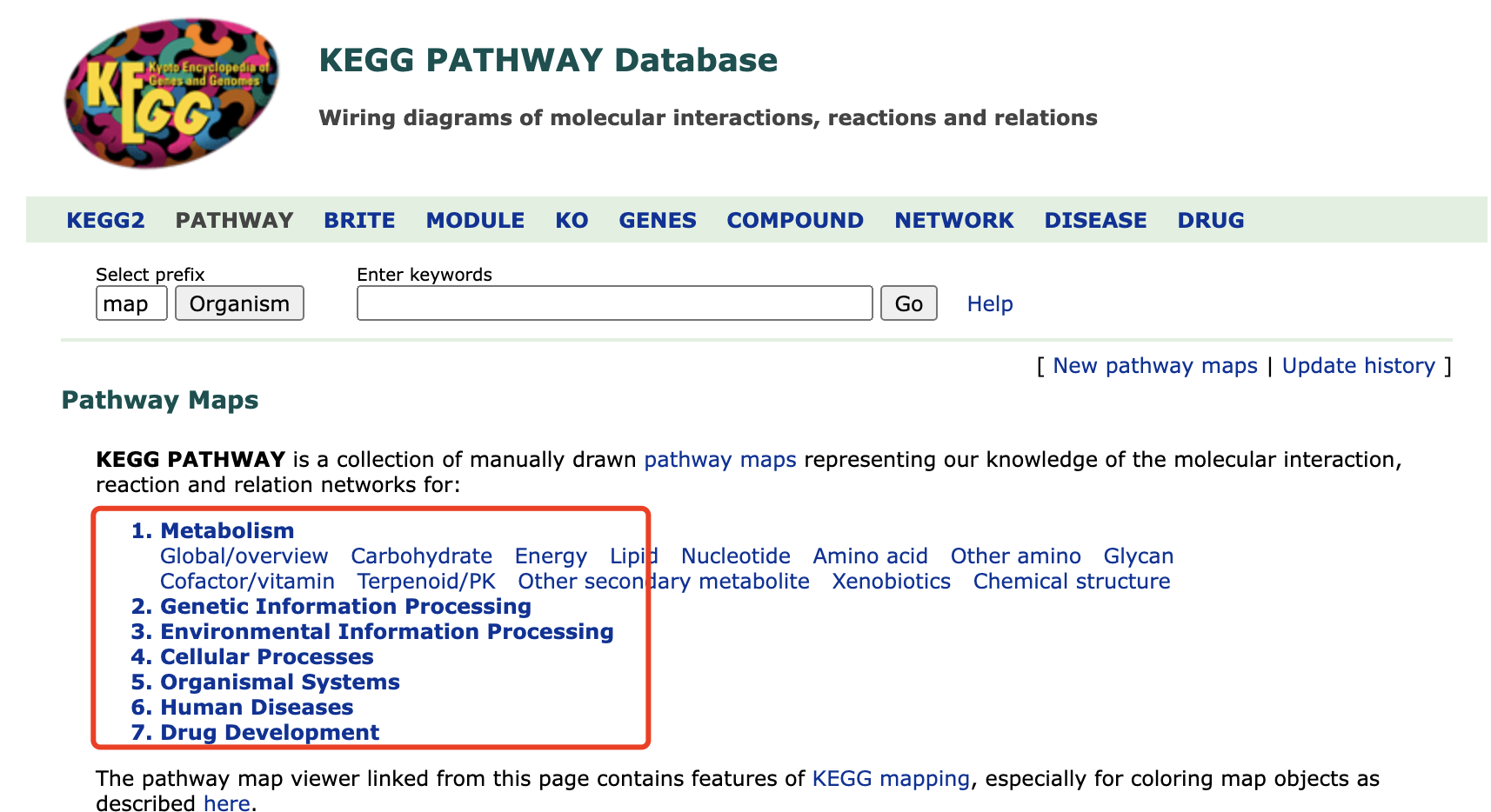
PATHWAY数据库中的通路被分为7类:
- 细胞过程(Cellular Processes)
- 环境信息处理(Environmental Information Processing)
- 遗传信息处理(Genetic Information Processing)
- 人类疾病(Human Diseases)
- 新陈代谢(Metabolism)
- 生物体系统(Organismal Systems)
- 药物相关(Drug Development)
对于人类(Homo sapiens)这个物种,KEGG数据库提供了物种特异性的通路,这些通路被详细地分类和注释。例如,在KEGG PATHWAY数据库中,可以通过特定的物种名称缩写(如hsa代表人类)来检索人类的特定通路,如hsa00010代表人类的糖酵解/糖异生通路。
少量基因做KEGG PATHWAY数据库注释
如果只有少量基因,其实每个基因去查询一下,就可以看到它的KEGG PATHWAY数据库注释信息啦, 每个基因都是独立的查询即可。比如大名鼎鼎的TP53,TP53基因大家都有所耳闻,而且也大概都知道它是 tumor protein p53的简称,其实它还有很多别名,比如BCC7;LFS1;P53;TRP53; :
可以看到其实是可以做很多数据库注释,但是绝大部分人只知道KEGG PATHWAY数据库。
如果仅仅是想做KEGG PATHWAY数据库注释,也是可以使用r代码的,借助这个 library(org.Hs.eg.db) 包即可:
rm(list=ls())
library(org.Hs.eg.db)
eg2symbol=toTable(org.Hs.egSYMBOL)
eg2name=toTable(org.Hs.egGENENAME)
eg2alias=toTable(org.Hs.egALIAS2EG)
eg2alis_list=lapply(split(eg2alias,eg2alias$gene_id),function(x){paste0(x[,2],collapse = ";")})
GeneList=mappedLkeys(org.Hs.egSYMBOL)
GeneList[1]
if( GeneList[1] %in% eg2symbol$symbol ){
symbols=GeneList
geneIds=eg2symbol[match(symbols,eg2symbol$symbol),'gene_id']
}else{
geneIds=GeneList
symbols=eg2symbol[match(geneIds,eg2symbol$gene_id),'symbol']
}
geneNames=eg2name[match(geneIds,eg2name$gene_id),'gene_name']
geneAlias=sapply(geneIds,function(x){ifelse(is.null(eg2alis_list[[x]]),"no_alias",eg2alis_list[[x]])})
借助于 org.Hs.eg.db 包,我们已经拿到了全部的人类全部基因的全名和别名,就是如下所示的4个变量:
- geneIds
- symbols
- geneNames
- geneAlias
同理,任意基因都可以去查询在 org.Hs.eg.db 包里面的有的信息:
> ls("package:org.Hs.eg.db")
[1] "org.Hs.eg" "org.Hs.eg_dbconn" "org.Hs.eg_dbfile"
[4] "org.Hs.eg_dbInfo" "org.Hs.eg_dbschema" "org.Hs.eg.db"
[7] "org.Hs.egACCNUM" "org.Hs.egACCNUM2EG" "org.Hs.egALIAS2EG"
[10] "org.Hs.egCHR" "org.Hs.egCHRLENGTHS" "org.Hs.egCHRLOC"
[13] "org.Hs.egCHRLOCEND" "org.Hs.egENSEMBL" "org.Hs.egENSEMBL2EG"
[16] "org.Hs.egENSEMBLPROT" "org.Hs.egENSEMBLPROT2EG" "org.Hs.egENSEMBLTRANS"
[19] "org.Hs.egENSEMBLTRANS2EG" "org.Hs.egENZYME" "org.Hs.egENZYME2EG"
[22] "org.Hs.egGENENAME" "org.Hs.egGENETYPE" "org.Hs.egGO"
[25] "org.Hs.egGO2ALLEGS" "org.Hs.egGO2EG" "org.Hs.egMAP"
[28] "org.Hs.egMAP2EG" "org.Hs.egMAPCOUNTS" "org.Hs.egOMIM"
[31] "org.Hs.egOMIM2EG" "org.Hs.egORGANISM" "org.Hs.egPATH"
[34] "org.Hs.egPATH2EG" "org.Hs.egPFAM" "org.Hs.egPMID"
[37] "org.Hs.egPMID2EG" "org.Hs.egPROSITE" "org.Hs.egREFSEQ"
[40] "org.Hs.egREFSEQ2EG" "org.Hs.egSYMBOL" "org.Hs.egSYMBOL2EG"
[43] "org.Hs.egUCSCKG" "org.Hs.egUNIPROT"
单个基因或者多个基因都可以通过代码,一个个的去注释,但是很浪费时间,因为如果基因很少量,大概率上大家 就直接使用网页工具了。
多个基因做注释需要统计学(超几何分布检验)
如果是少量的几个基因,确实是可以一个个去网页工具里面查询获取具体的信息,但是如果是几十个或者上百个基因,比如我们做完差异分析后的基因列表,就需要借助统计学(超几何分布检验),在KEGG数据库注释中使用统计学方法,尤其是超几何分布检验,是为了识别差异基因集中富集的生物学通路。以下是该过程的一般步骤:
-
差异基因集的获取:首先,你需要从一个实验条件(如疾病状态)与另一个实验条件(如正常状态)的比较中获得一组差异表达的基因。
-
基因功能注释:将这些差异基因与KEGG数据库中的通路进行匹配。这通常涉及到将基因标识符(如Ensembl ID、GenBank Accession号码等)转换为KEGG Orthology (KO)编号。
-
超几何分布检验:利用超几何分布检验来确定某个通路中的基因在差异基因集中是否显著富集。超几何分布检验的基本原理是评估从特定大小的群体(背景基因集)中随机抽取一定数量的样本(前景基因集,即差异基因集)时,获得特定特征(如属于某个KEGG通路)的基因数目的概率。
检验的统计公式通常为:
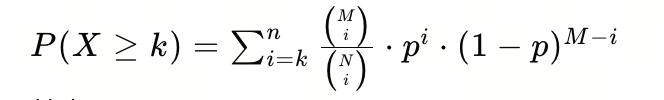
其中:
- ( N ) 是背景基因集中所有基因的总数。
- ( M ) 是背景基因集中具有某种特定功能的基因数。
- ( n ) 是前景基因集中的基因数。
- ( k ) 是前景基因集中具有该特定功能的基因数。
- ( p ) 是在背景基因集中抽取到具有该功能基因的概率,( p = \frac{M}{N} )。
-
多重检验校正:由于一次分析中可能涉及多个通路的检验,因此需要使用如Bonferroni校正或False Discovery Rate (FDR)等方法来校正P值,以减少假阳性结果。
-
结果解释:最后,根据校正后的P值确定哪些通路是显著富集的,并据此解释差异基因集中的生物学功能。
-
可视化:使用图表或富集图(如气泡图、条形图)来直观展示富集分析的结果。
这个过程涉及到对统计学原理的深刻理解以及对KEGG数据库结构的熟悉。在实际应用中,许多生物信息学工具和软件包(如R包clusterProfiler)可以自动化这些步骤,简化富集分析的过程。
当然了,看不懂上面的统计学文字描述很正常, 你可以试试看朴素的理解它,当然可以。超几何分布检验可以用一个更朴素的方式来理解,通过一个简单的例子来说明。
假设你有一个装满球的箱子,其中一些球是红色的,其余的是蓝色的。红色球和蓝色球的比例代表了整个群体(也就是背景)中特定特征(比如红色)的比例。现在,你闭上眼睛从箱子里随机取出几个球,组成一个较小的样本组(也就是前景)。你感兴趣的是,取出的样本组中红色球的比例是否显著高于整个箱子中红色球的比例。
超几何分布检验的朴素理解
-
随机抽取:你从箱子里随机抽取球(不把抽出来的球放回去),看能抽到多少红球。
-
期望比例:整个箱子中红球和蓝球的比例给你一个期望值,也就是在随机抽取的情况下,你期望抽到的红球的比例。
-
实际观察:你实际抽出来的样本中红球的比例与你的期望值进行比较。
-
检验统计量:超几何分布检验会计算在随机情况下,抽取的样本中红球比例至少和你实际观察到的一样高(或更高)的概率。
-
显著性:如果你观察到的红球比例远高于期望值,并且这种情况发生的概率非常低,那么你可能得出结论:抽取的样本中红球是“显著富集”的。
应用到KEGG富集分析
在KEGG富集分析中,这个例子可以这样对应:
-
箱子和球:箱子代表你研究的生物体的所有基因(背景基因集),球代表这些基因。红色球代表参与某个特定生物学过程或通路的基因。
-
抽取样本:你从箱子中抽取的球代表你通过实验发现的差异基因集(前景基因集)。
-
期望与观察:你期望前景基因集中参与特定通路的基因比例与背景基因集中的比例相似。但如果你观察到前景基因集中该通路的基因显著更多,这可能意味着该通路在你的实验条件下起到了特别的作用。
-
超几何分布检验:通过这个检验,你可以统计上判断前景基因集中某个通路的富集是随机发生的,还是很可能反映了真实的生物学现象。
当然了,如果你尝试去理解这个故事本身,也很难朴素的理解超几何分布检验,另外一个比较好的方式就是通过图像来展示,比如下面的韦恩图 ;
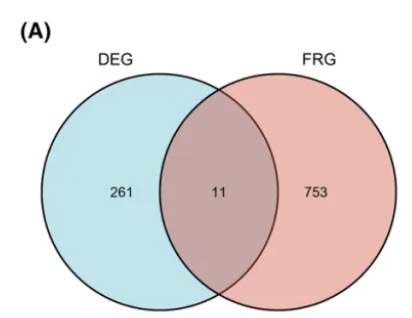
其中的deg就是你需要注释的基因列表,通常是几十个甚至上百个,然后FRG就是kegg等数据库里面的一个pathway啦,上面的韦恩图(Venn diagram)展现了两个基因列表的交集的数量,就可以通过超几何分布检验判定这个交集数量是否有统计学显著性。
我们通常是不需要一个个去手工整理每个pathway的基因列表,然后一个个去跟我们的需要注释的基因列表去做超几何分布检验的, 因为在代码里面都是批量而且自动化的!如下所示的代码就是一个很好的例子:
library(clusterProfiler)
library(ggplot2)
data(geneList, package='DOSE')
de <- names(geneList)[1:100]
yy <- enrichKEGG(de, pvalueCutoff=0.05)
head(yy@result[,1:3])
dotplot(yy)
如果是初次接触r语言,可能会对上面的代码比较困惑。其实就是使用了clusterProfiler包来进行KEGG富集分析,并且利用ggplot2包来生成可视化图形。下面是对每一行代码的详细解释:
library(clusterProfiler)
这行代码加载了clusterProfiler包,这是一个用于生物数据富集分析的R包,可以进行基因本体(GO)和KEGG通路富集分析。library(ggplot2)
加载ggplot2包,这是一个流行的R包,用于创建高质量的统计图形。data(geneList, package='DOSE')
使用data()函数从名为DOSE的R包中加载名为geneList的数据集。DOSE包是一个包含多个用于富集分析的数据集的包。de <- names(geneList)[1:100]
这行代码从geneList数据集中提取前100个基因的名称,并将它们存储在名为de的变量中。names(geneList)获取geneList中所有基因的名称,[1:100]选取前100个。yy <- enrichKEGG(de, pvalueCutoff=0.05)
使用enrichKEGG函数进行KEGG富集分析。该函数的输入是基因列表de,以及一个阈值pvalueCutoff,这里设置为0.05。这意味着只有校正后的P值(通过多重假设检验校正)小于0.05的通路才会被认为在富集分析中是显著的。yy变量存储了富集分析的结果。head(yy@result[,1:3])
这行代码查看yy对象中的结果result的前几行数据。yy@result访问yy对象的result槽,它包含了富集分析的详细结果。head()函数默认显示前6行,但通过[,1:3]选取了前3列的数据,即通路ID、通路名称和校正后的P值。dotplot(yy)
使用dotplot()函数生成一个点图,这是一种常用于展示富集分析结果的图形。该图将展示每个显著富集的通路,以及与之相关的基因数量和显著性水平。
如下所示的气泡图展示富集分析结果:
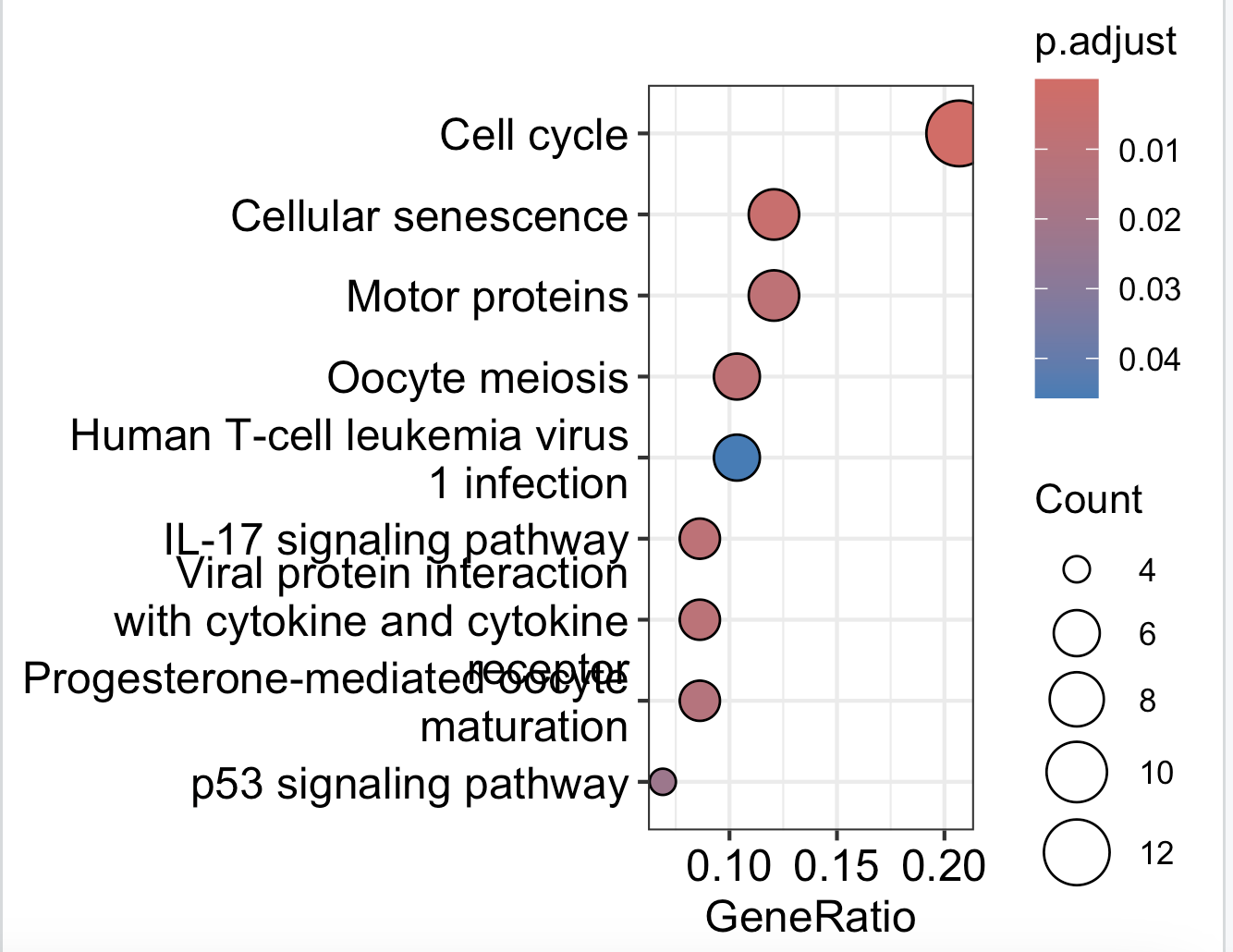
这个结果其实对应的是一个表格,里面有更多更丰富的信息,但是我们的气泡图默认就展现排名靠前的通路而已。而且里面的每一个通路其实又可以通过一个韦恩图来可视化。上面的 cell cycle通路之所以被显著的富集,其实就是cell cycle通路和我们的差异基因交集太多了,两个基因列表的韦恩图交集过多就容易达到统计学显著性。
给y叔打call
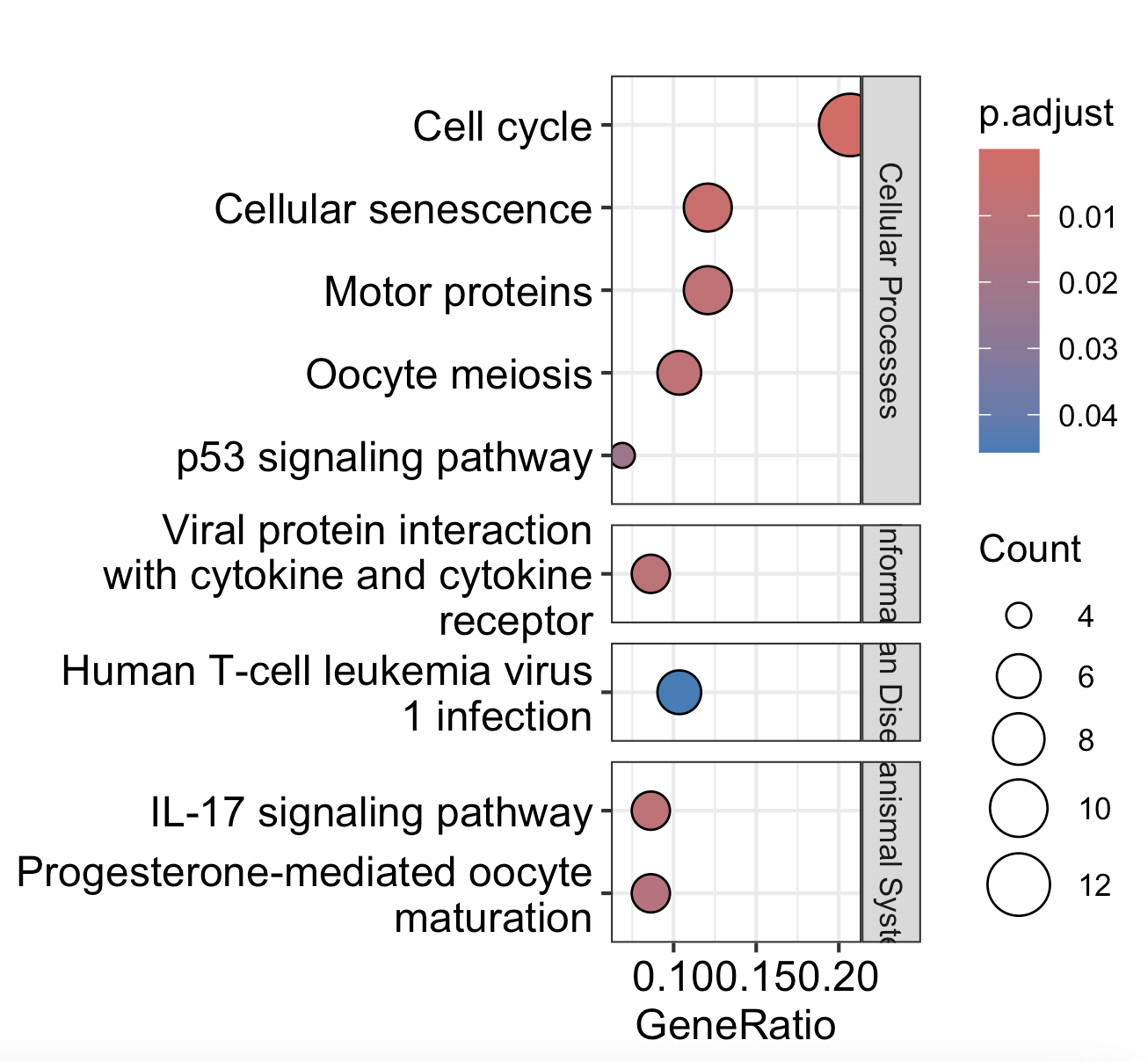
终于回到了我们的主题,把kegg注释结果图分门别类,如果你还是比较早期的clusterProfiler包是没办法做到。因为y叔是在最新的clusterProfiler包里面加上了这个kegg分门别类的信息:
> as.data.frame(sort(table(yy@result$category)))
Var1 Freq
1 Genetic Information Processing 3
2 Cellular Processes 11
3 Metabolism 14
4 Environmental Information Processing 19
5 Organismal Systems 35
6 Human Diseases 41
> head(yy@result[,1:2])
category subcategory
hsa04110 Cellular Processes Cell growth and death
hsa04218 Cellular Processes Cell growth and death
hsa04814 Cellular Processes Cell motility
hsa04114 Cellular Processes Cell growth and death
hsa04657 Organismal Systems Immune system
然后我们微信交流群的王晶(中国科学技术大学)同学写了下面的代码比较好的可视化了这个kegg注释结果图分门别类
dotplot(yy)+facet_grid(rows = vars(category),scales = 'free_y',space = 'free_y')+
scale_color_gradientn(colors = c('#BF1E27','#FEB466','#F9FCCB','#6296C5','#38489D'))
效果如下:
后记
可以看到,生物信息学数据分析里面,代码确实是很重要,因为你要个性化实现很多细致的需求,但是背景知识也不容小觑,如果你不了解kegg数据库是什么不知道什么是超几何分布检验,也不知道kegg数据库的pathway是可以分门别类的,那么代码再牛其实也很难有所作为,无非就是你导师手下吭哧吭哧写代码干活的,永永远远都没办法有自己的想法。