Smart-seq2和10x这两个单细胞技术是现在初学者进入单细胞领域最需要掌握的,它们代表着单细胞的两个全然不同的发展策略。
绝大部分的技术原理介绍会从 单细胞悬浮液制备到测序细节面面俱到,其实并不那么的初学者友好。给大家推荐了一个高度精炼的综述,这个综述于2020年9月发表在 《Experimental & Molecular Medicine》杂志,标题是:《Single-cell sequencing techniques from individual to multiomics analyses》,链接是:https://www.nature.com/articles/s12276-020-00499-2
- 首先呢,smart-seq2技术依赖于C1这个仪器,每次都是96个细胞一起测序,每个细胞的测序量这个综述可能是写错了,应该是1M-10M为佳,不太可能是100-1000个M,最重要的是它可以覆盖到整个RNA分子的全长测序,每个细胞都是独立的测序,独立的fastq文件哦 。
- 然后呢,对于10X技术单细胞转录组呢,每次可以测好几千的细胞,每个细胞只需要5-10K的reads,而且仅仅是测RNA分子的一段即可,全部的细胞都混合在一起是一个fastq文件,虽然说有barcode可以区分,可以拆分成为不同细胞的表达量矩阵。
正常情况下,大家只需要按需选择10x或者smart-seq2技术平台做单细胞转录组数据即可,但现在smart-seq2这样的细胞通量很低的技术已经式微了,基本上大家都会选择细胞通量高的平台,尤其是10X单细胞转录组技术。不过大家很可能会被BD技术平台的销售也吸引一下,陷入选择困难症,因为都细胞通量很高!
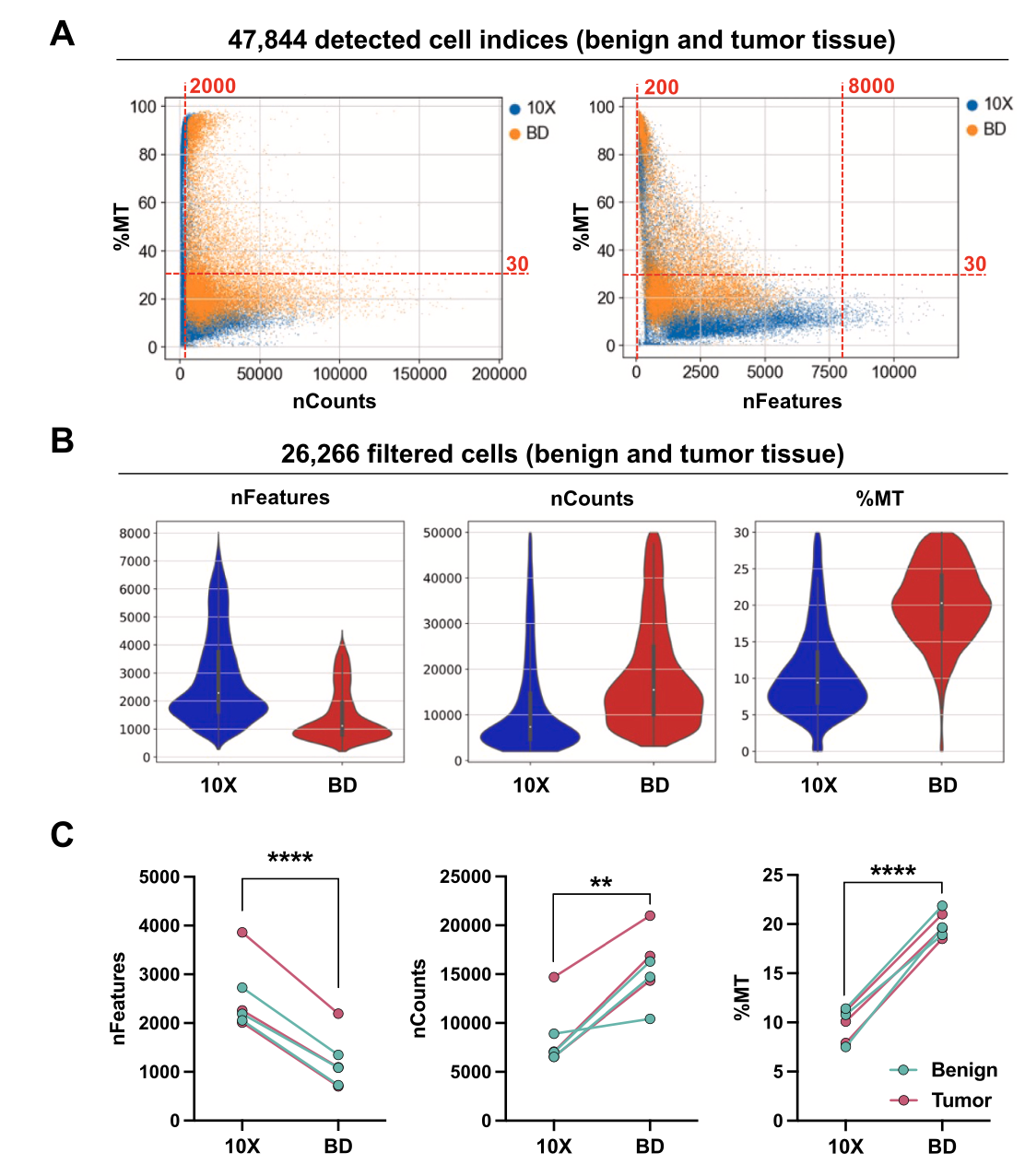
最近在群里看到了一个2024的测评文章:《Comparative analysis of 10X Chromium vs. BD Rhapsody whole transcriptome single-cell sequencing technologies in complex human tissues》,基本上的结论是10X单细胞转录组全方位吊打BD平台。线粒体含量问题
首先是BD平台的线粒体含量过高:
单细胞转录组测序中,如果观察到每个细胞的线粒体含量(即线粒体基因表达水平)过高,可能会有以下几种影响和解释:
- 细胞代谢活性高:
- 线粒体是细胞的能量工厂,负责产生大部分的细胞能量(ATP)。线粒体基因表达水平高可能表明细胞的代谢活性增强。
- 细胞类型特征:
- 某些细胞类型,如肌肉细胞,具有较高的线粒体含量,这是它们的正常生理特征。
- 细胞应激反应:
- 线粒体基因表达的增加可能是细胞对应激条件(如缺氧、营养缺乏等)的响应。
- 疾病状态指示:
- 在某些疾病状态下,如某些类型的癌症,肿瘤细胞可能会展示出较高的线粒体活性,以支持它们的快速增殖。
- 技术偏差:
- 高线粒体含量可能是由于测序或样本处理过程中的技术偏差。例如,基因组DNA的污染可能导致线粒体DNA被过度测序。
- 数据解释复杂性增加:
- 高线粒体基因表达可能会影响对细胞其他基因表达模式的分析和解释,因为线粒体序列在数据分析中可能需要特别处理。
- 细胞死亡和凋亡:
- 在某些情况下,线粒体基因表达的增加可能与细胞死亡和凋亡过程有关。
- 细胞分化状态:
- 线粒体活性的变化可能与细胞的分化状态有关,不同分化阶段的细胞可能有不同的能量需求和线粒体活性。
- 数据分析挑战:
- 高线粒体含量可能需要在数据分析时进行特别的考虑,比如在数据标准化和基因表达量估算时排除线粒体基因的影响。
- 研究假设检验:
- 高线粒体含量可能提示研究者检验与线粒体功能相关的生物学假设,如能量代谢、氧化应激等。
在分析单细胞转录组数据时,研究者需要考虑线粒体基因表达水平的生物学意义和潜在的技术因素,以确保数据解释的准确性。如果必要,可能需要采取额外的步骤来校正线粒体基因的过度表达,以便更准确地分析细胞核基因的表达模式。单个细胞检测到的基因数量问题
可以看到,具体到每个单细胞亚群,都是10x平台检测到的基因数量远多于bd平台哦:
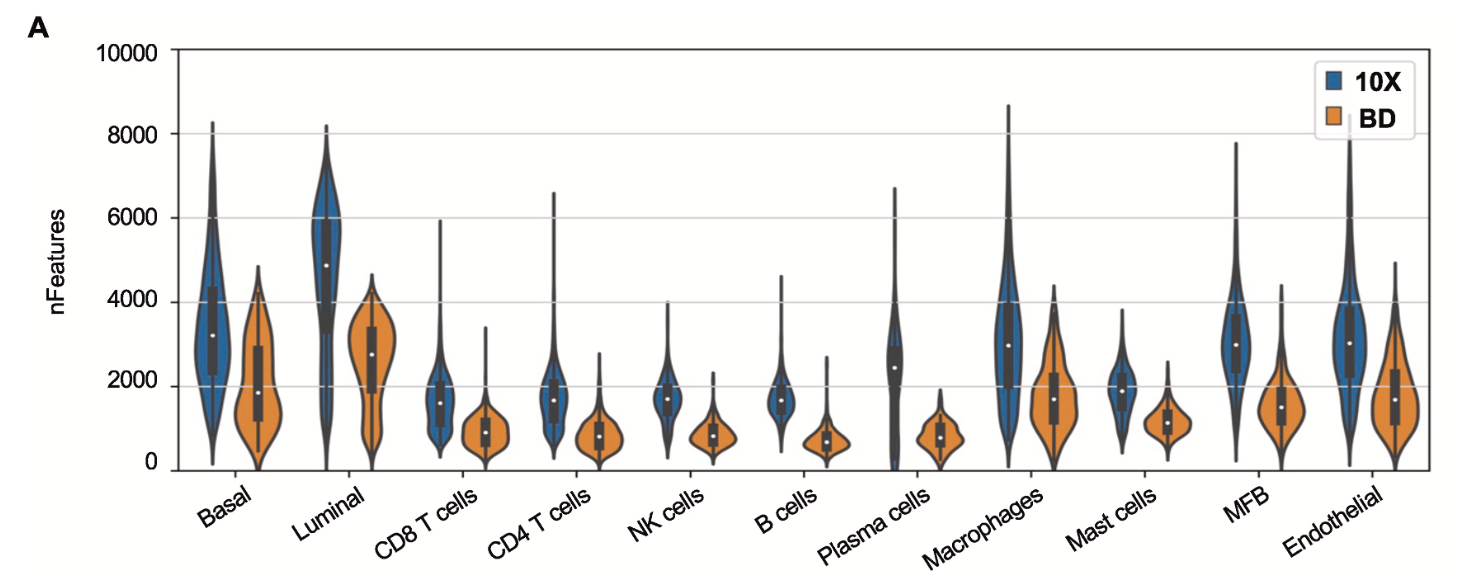
在单细胞转录组测序中,每个细胞检测到的基因数量较高可能带来以下好处:
- 高线粒体含量可能提示研究者检验与线粒体功能相关的生物学假设,如能量代谢、氧化应激等。
- 提高细胞类型分辨率:
- 检测到更多基因有助于更准确地区分不同的细胞类型和状态,因为每个细胞类型的基因表达谱是独特的。
- 增强异质性识别:
- 高基因检测数量有助于揭示细胞群体内部的异质性,即使是在看似相同的细胞群体中也可能存在不同的亚群。
- 改善功能分析:
- 检测到的基因越多,对细胞功能状态的推断就越准确,有助于理解细胞在生物学过程或疾病中的作用。
- 促进生物标志物发现:
- 更多的基因表达信息有助于识别潜在的生物标志物,这些标志物可以用于疾病诊断或治疗反应的预测。
- 支持复杂生物学过程的研究:
- 高基因检测数量有助于研究复杂的生物学过程,如细胞分化、发育和疾病进程。
- 提高数据的可靠性:
- 检测到的基因数量越多,数据集通常被认为越可靠,减少了由于技术变异或低表达基因遗漏带来的偏差。
- 增强多变量分析能力:
- 在进行聚类分析、主成分分析(PCA)等多变量统计分析时,更多的基因可以提供更丰富的数据维度。
- 改善基因网络和通路分析:
- 检测到的基因数量多,有助于构建和分析基因之间的相互作用网络和信号通路。
- 有助于罕见事件的检测:
- 某些生物学事件可能只涉及少数细胞,高基因检测数量有助于捕捉这些罕见事件。
- 提供更多的数据整合机会:
- 高基因覆盖率为不同样本或实验的数据整合提供了更多可能性,有助于跨研究比较和数据集的合并分析。
然而,值得注意的是,每个细胞检测到的基因数量过高也可能带来一些挑战,如增加数据分析的复杂性、需要更大的计算资源和存储空间,以及可能需要更精细的数据预处理和标准化方法。因此,研究者需要在实验设计和数据分析时权衡这些因素,以确保结果的准确性和可解释性。让我们重新分析这个数据
大家可以打开 https://zenodo.org/records/8063560 就能下载到里面的 Prostate_RAW.h5ad 文件,约677M的文件。很容易读取进去走Seurat流程,代码如下所示:
#BiocManager::install("zellkonverter") library(zellkonverter) ad <- readH5AD('Prostate_RAW.h5ad') adata_Seurat <- as.Seurat(ad, counts = "X", data = NULL) sce.all = CreateSeuratObject( counts = adata_Seurat@assays$originalexp ) as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2]) head(sce.all@meta.data, 10) table(sce.all$orig.ident) library(stringr) phe=str_split(colnames(sce.all),'[-_]',simplify = T) table(phe[,2]) phe=adata_Seurat@meta.data sce.all$orig.ident=phe$sample如下所示,可以看到具体的每个样品在每个平台下面的降维聚类分群的区别:
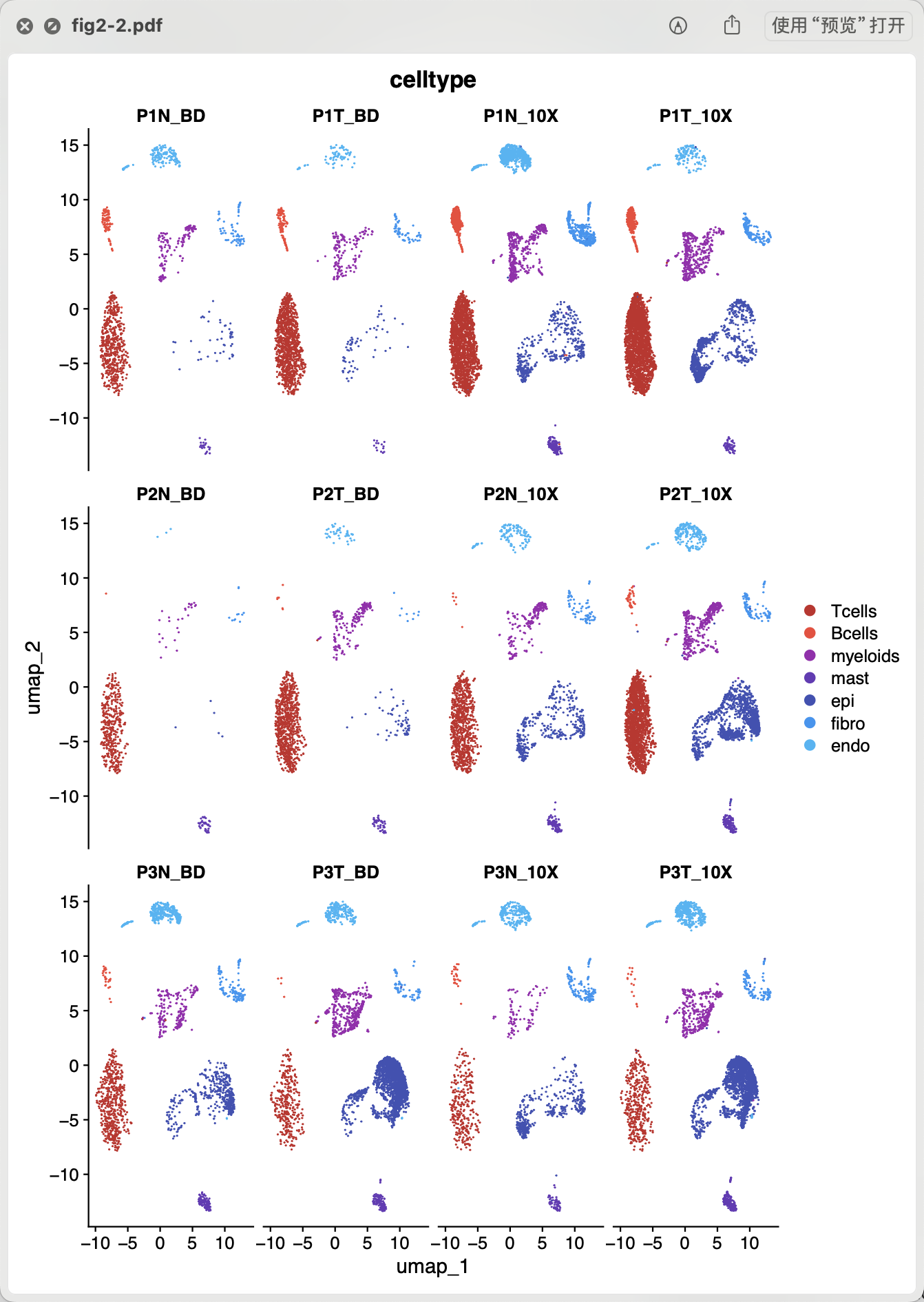
- 高基因覆盖率为不同样本或实验的数据整合提供了更多可能性,有助于跨研究比较和数据集的合并分析。