之前我们介绍过irGSEA:基于秩次的单细胞基因集富集分析整合框架,针对17种常见的Functional Class Scoring (FCS)方法在单细胞转录组里面进行了benchmark,感兴趣的可以仔细读一下。
我们首先了解一下打分工具的本质,首先是带有生物学功能意义的基因集合的数据库资源,其次是统计学公式。前者可以是免疫或者代谢等基因集,主要是来源于msigdb等数据库。后者统计学公式对大家来说最容易理解的就是gsea或者gsva啦。比如我前两天介绍的:单细胞GSVA分析专用R包
今天我们分享一个工具 | TCellSI:首个全面评估T细胞状态的创新工具,是2024年6月21日,四川大学华西医院郭安源教授团队首次开发了一种能够系统地评估T细胞状态的新工具,TCellSI (T Cell State Identifier)。
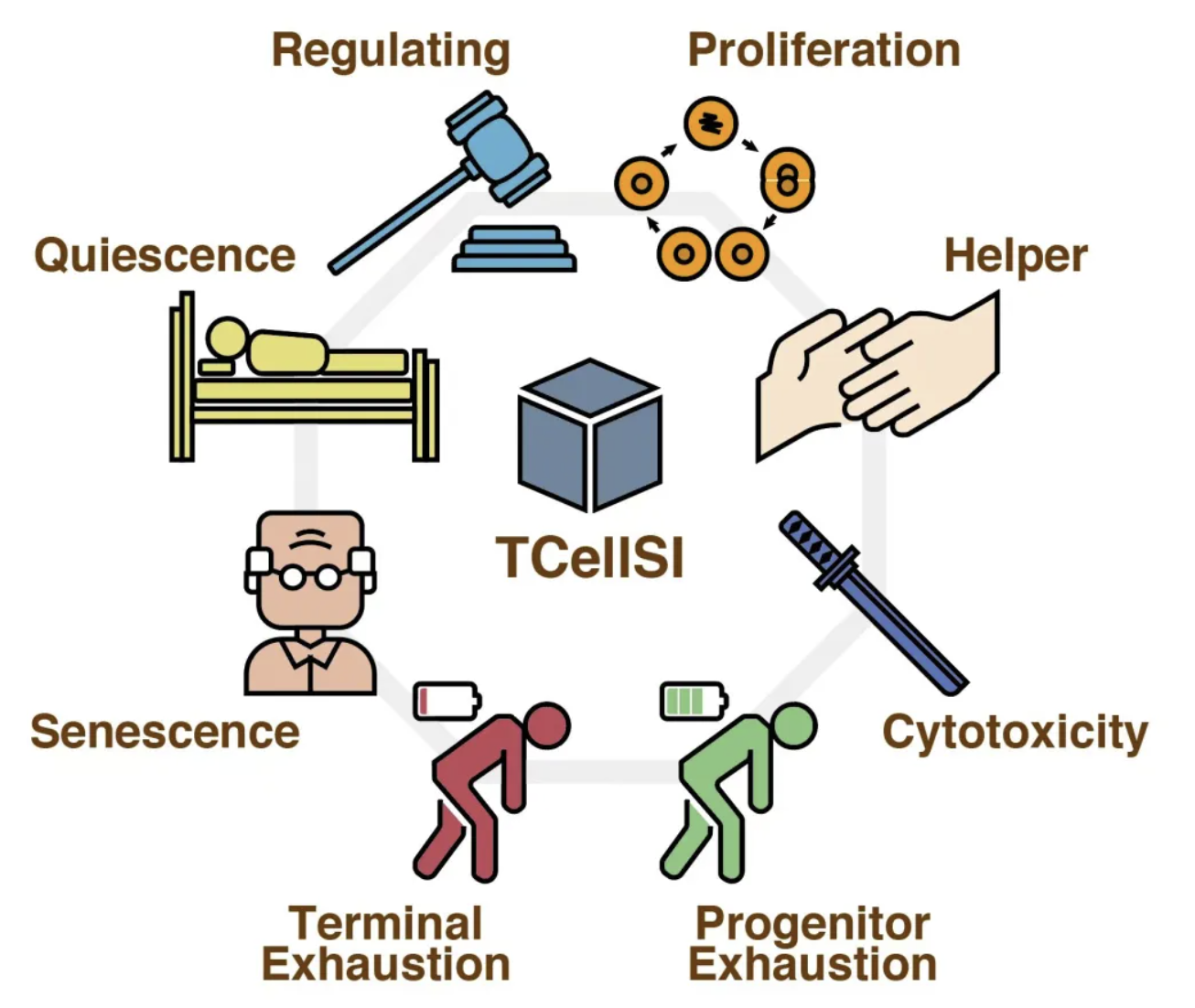
目前准确评估T细胞的八种不同状态,这些状态包括:
● 静止(Quiescence)
● 调节(Regulating)
● 增殖(Proliferation)
● 辅助(Helper)
● 细胞毒性(Cytotoxicity)
● 初始耗竭(Progenitor exhaustion)
● 终末耗竭(Terminal exhaustion)
● 衰老(Senescence)
这些状态的评估结果被称为T细胞状态评分(TCSS, T Cell State Score)。通过这些评分,研究人员可以深入了解T细胞在免疫反应中的具体功能和角色。
TCellSI借助Mann-Whitney U统计量,通过特定的标记基因集和参考谱为样本进行T细胞状态的评估。TCellSI在开发和验证过程中,涵盖了大规模数据,包括来自20个数据集的 379个 T 细胞系、34,730个单细胞、4,477个pseudo-bulk样本、33 种癌症类型的 10,535 名癌症患者、20种组织类型的 7,862个正常样本、免疫治疗队列中的 674个样本,以及 884个病毒感染的非癌症外周血样本。TCellSI不仅能准确描述T细胞状态,而且在免疫环境中显现出高度的预测价值。
先看看包的帮助文档
安装和加载:
# install.packages("devtools")
devtools::install_github("GuoBioinfoLab/TCellSI")
library(TCellSI)
然后一步到位就可以评估T细胞的八种状态:
## 示例表达数据
## 要求是完整的基因表达矩阵(log2转换后的RNA-seq TPM标准化基因表达矩阵)
sample_expression <- TCellSI::exampleSample
sample_expression[1:10, ]
# 如下所示的示例数据表达量矩阵格式:
## SRR5088825 SRR5088828 SRR5088830 SRR5088833 SRR5088835
## 5_8S_rRNA 0.000000 0.000000 0.0000000 0.0000000 0.000000
## 5S_rRNA 0.000000 0.000000 0.4346853 0.0000000 0.000000
## 7SK 0.000000 0.000000 0.0000000 0.0000000 0.000000
## A1BG 1.907599 3.284418 4.0821248 3.2864715 2.863233
## A1BG-AS1 3.083914 2.021561 4.5169002 2.9347126 2.918992
## A1CF 0.000000 0.000000 0.0000000 0.0000000 0.000000
## A2M 9.812037 9.516751 5.7541683 6.9308912 7.394556
## A2M-AS1 2.107557 1.889085 0.1596072 0.8729146 1.206896
## A2ML1 0.000000 0.000000 0.0000000 0.0000000 0.000000
## A2ML1-AS1 0.000000 0.000000 0.0000000 0.0000000 0.000000
## 进行TCSS的计算,TCSS是0-1之间的数值
ResultScores_TCSS <- TCSS_Calculate(sample_expression)
返回的就是每个样品的8种打分值:
ResultScores_TCSS
## SRR5088825 SRR5088828 SRR5088830 SRR5088833 SRR5088835
## Quiescence 0.6422158 0.6153140 0.5105856 0.4520591 0.6951249
## Regulating 0.4404173 0.3974391 0.1670356 0.2615326 0.5454356
## Proliferation 0.5780474 0.5761891 0.3994404 0.4282601 0.6184267
## Helper 0.5647604 0.4063729 0.2121070 0.1735861 0.6547372
## Cytotoxicity 0.6482720 0.5826073 0.1829111 0.2358969 0.5888452
## Progenitor_exhaustion 0.5905679 0.5308304 0.2482848 0.2461412 0.6199635
## Terminal_exhaustion 0.6624943 0.6222188 0.3311532 0.3387099 0.6042142
## Senescence 0.5921543 0.5725185 0.2896508 0.3206251 0.6043355
对于这个打分结果矩阵,可以有很多可视化方法啦, 具体可以看 R包 的官方文档 (https://github.com/GuoBioinfoLab/TCellSI/)
核心之一:人工整理的基因列表:
> markers
$Quiescence
[1] "BTG2" "KLF2" "FOXP1" "RUNX1" "MYC"
$Regulating
[1] "TNFRSF9" "TNFRSF18" "ENTPD1" "IKZF4" "LRRC32" "IKZF2"
[7] "STAT5A" "FOXP3" "CD4"
$Proliferation
[1] "MKI67" "TYMS" "TTK" "CKS1B" "BRIP1" "GINS2" "CKS2" "ANLN"
[9] "CD4" "CD8A" "CD8B" "CD3G" "CD3D" "CD3E" "CD247"
$Helper1_2
[1] "LTA" "TNF" "IL2" "IL18" "TBX21" "LTBR"
[7] "CXCR3" "IFNG" "KLRD1" "HAVCR2" "DPP4" "TNFSF11"
[13] "CCR5" "IFNGR1" "STAT1" "STAT4" "CCR4" "CCR6"
[19] "GZMK" "IFNGR1" "PARP8" "PVRIG" "SLC4A10"
$Helper17
[1] "TGFB1" "STAT3" "RORA" "RORC" "IL22" "IL21"
[7] "IL17F" "IL17A" "CCR6" "GZMK" "IFNGR1" "PARP8"
[13] "PVRIG" "SLC4A10"
$Fhelper
[1] "STAT3" "ICOS" "CXCR5" "BCL6" "CXCL13" "PDCD1"
[7] "BATF" "CXCR3" "KLRB1" "LTA" "MAP4K1" "CCR4"
[13] "CCR6" "GZMK" "IFNGR1" "PARP8" "PVRIG" "SLC4A10"
$Cytotoxicity
[1] "GZMB" "GZMH" "GZMK" "GZMA" "TIA1" "PRF1" "LAMP1"
[8] "GNLY" "FASLG" "SLAMF7" "ZAP70" "CD69" "TNF"
$Progenitor_exhaustion
[1] "CTLA4" "PDCD1" "HNF1A" "SLAMF6" "CD8A" "CD8B"
[7] "TNF" "ICOS" "TNFSF14" "SATB1" "EOMES" "ID3"
[13] "TCF7" "CXCR3" "GZMK" "SELL" "IL7R" "CCR7"
[19] "CXCL10" "CD44" "BCL6" "CD27" "CD28" "CD38"
$Terminal_exhaustion
[1] "CXCL13" "ENTPD1" "PRF1" "GZMH" "GZMB" "GZMA"
[7] "BATF" "LYST" "BATF" "TIGIT" "LAG3" "HAVCR2"
[13] "PDCD1" "PRDM1" "RBPJ" "SLAMF7" "CCL4" "TNFSF10"
[19] "TBX21" "NFATC1" "HIF1A" "TNFRSF4" "CXCR6" "PTPN7"
[25] "EWSR1" "HLA-DPB1" "HLA-DPA1" "UBXN1" "PSMB9" "LY6E"
[31] "CCNDBP1" "IRF9" "LCP2"
$Senescence
[1] "CD3G" "CD3D" "CD3E" "CD247" "KLRG1" "B3GAT1" "TP53"
[8] "MAPK14" "MAPK8" "MAPK1" "CDKN2A"
核心之二:
打分算法:
感兴趣可以让人工智能大模型帮你解读一下这两个代码文件:
81 R/CSS_Calculate.R
112 R/TCSS_Calculate.R
每个代码里面的内容并不多哈,相信大家可以啃下去的!
应用于小规模单细胞表达量矩阵
很容易从Seurat对象里面的拿到了单细胞表达量矩阵,一般来说都是两三万个基因然后几万个甚至几十万细胞数量。所以这个时候计算资源消耗比较大而且很慢,而且单细胞表达量矩阵里面的drop-out非常严重,计算会不准确,下面的两个方法都可以获取具体的表达量矩阵哈:
GetAssayData(sce , slot='counts',assay='RNA')
sce@assays$RNA$counts
推荐使用伪bulk表达量矩阵方法
比如下面的案例是首先对自己的单细胞表达量矩阵进行降维聚类分群后并且生物学注释,这样的话每个细胞就有了celltype属性,然后多个样品可以分开,如下所示:
# v4 是 AverageExpression
av <-AggregateExpression(sce.all ,
group.by = c("orig.ident","celltype"),
assays = "RNA")
av=as.data.frame(av[[1]])
head(av) # 可以看到是整数矩阵
#write.csv(av,file = 'AverageExpression-0.8.csv')
cg=names(tail(sort(apply(log(av+1), 1, sd)),1000))
pheatmap::pheatmap(cor(as.matrix(log(av[cg,]+1))))
save(av,file = 'av_for_pseudobulks.Rdata')
上面的 log(av+1) 矩阵,就可以直接使用TCSS_Calculate去计算啦。
其它打分推荐
之前我们介绍过irGSEA:基于秩次的单细胞基因集富集分析整合框架,针对17种常见的Functional Class Scoring (FCS)方法在单细胞转录组里面进行了benchmark,感兴趣的可以仔细读一下。使用方法也很简单,如果是真的全部的单细胞表达量矩阵就会很慢很慢哦,如下所示代码:
# 包括GSEA, GSVA,PLAGE, addModuleScore, SCSE, Vision, VAM, gficf, pagoda2, Sargent, AUCell、UCell、singscore、ssGSEA、JASMINE, Viper等等
# https://mp.weixin.qq.com/s/VI4ISO6y5_rt8Yy_VnIR-w
library(irGSEA)
# irGSEA通过内置的msigdbr包的基因列表进行打分
# #### Hallmark基因集打分 ####
sce.all <- irGSEA.score(object = sce.all, assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "H",
subcategory = NULL, geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea" ),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
可视化;
基本上就是Seurat包的5种可视化,单细胞数据分析里面最基础的就是降维聚类分群,参考前面的例子:人人都能学会的单细胞聚类分群注释 ,这个大家基本上问题不大了,使用seurat标准流程即可,不过它默认出图并不好看,详见以前我们做的投票:可视化单细胞亚群的标记基因的5个方法,下面的5个基础函数相信大家都是已经烂熟于心了:
- VlnPlot(pbmc, features = c(“MS4A1”, “CD79A”))
- FeaturePlot(pbmc, features = c(“MS4A1”, “CD79A”))
- RidgePlot(pbmc, features = c(“MS4A1”, “CD79A”), ncol = 1)
- DotPlot(pbmc, features = unique(features)) + RotatedAxis()
- DoHeatmap(subset(pbmc, downsample = 100), features = features, size = 3)
这个T细胞状态打分R包,就可以去跟我们的降维聚类分群结合起来进行5种可视化展示。