前面我在笔记:作者仅提供了fpkm格式表达量矩阵的转录组测序数据集该如何重新分析呢 提到了一个小技巧,是可以通过下面的r代码读取geo数据库里面的转录组测序表达量矩阵,这个矩阵来自于geo官方的转录组定量流程 ,如下所示的r代码 :
urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?format=file&type=rnaseq_counts"
path <- paste(urld, "acc=GSE182923", "file=GSE182923_raw_counts_GRCh38.p13_NCBI.tsv.gz", sep="&");
tbl <- as.matrix(data.table::fread(path, header=T, colClasses="integer"), rownames=1)
虽然说我们确实是在单细胞天地,生信菜鸟团,生信技能树等多个公众号转发了:作者仅提供了fpkm格式表达量矩阵的转录组测序数据集该如何重新分析呢 里面的小技巧,但仍然是各个交流群还是有人发问,关于转录组测序的公共数据集如何分析,因为大家看到的常规教程都是之前的表达量芯片的数据分析流程。
转录组测序首先是表达量矩阵是一个问题, 其次处理所用的r包,统计学方法也不一样。
比如:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE254592
对应的文章是:Programmed cell death-1 is involved with peripheral blood immune cell profiles in patients with hepatitis C virus antiviral therapy. PLoS One 2024;19(5):e0299424. PMID: 38781172
如果是看文件:
GSE254592_Group01_vs_Group02_cds_expression_diff_summary.txt.gz 2.2 Mb (ftp)(http) TXT
GSE254592_Group01_vs_Group03_cds_expression_diff_summary.txt.gz 2.2 Mb (ftp)(http) TXT
GSE254592_Group02_vs_Group04_cds_expression_diff_summary.txt.gz 2.2 Mb (ftp)(http) TXT
GSE254592_Group03_vs_Group04_cds_expression_diff_summary.txt.gz 2.2 Mb (ftp)(http) TXT
GSE254592_TR_2305.genes.fpkm_table_annotation.txt.gz 6.4 Mb (ftp)(http) TXT
GSE254592_merged.gtf.gz 15.5 Mb (ftp)(http) GTF
确实是一个fpkm格式的表达量矩阵,虽然说geo页面给出来了一下 差异分析的组合,但是没办法替代我们自己想从原始的counts矩阵开始的差异分析需求。
其实,如果是现在的大家仔细看说geo页面,就可以看到页面多了一个下载渠道的按钮 :
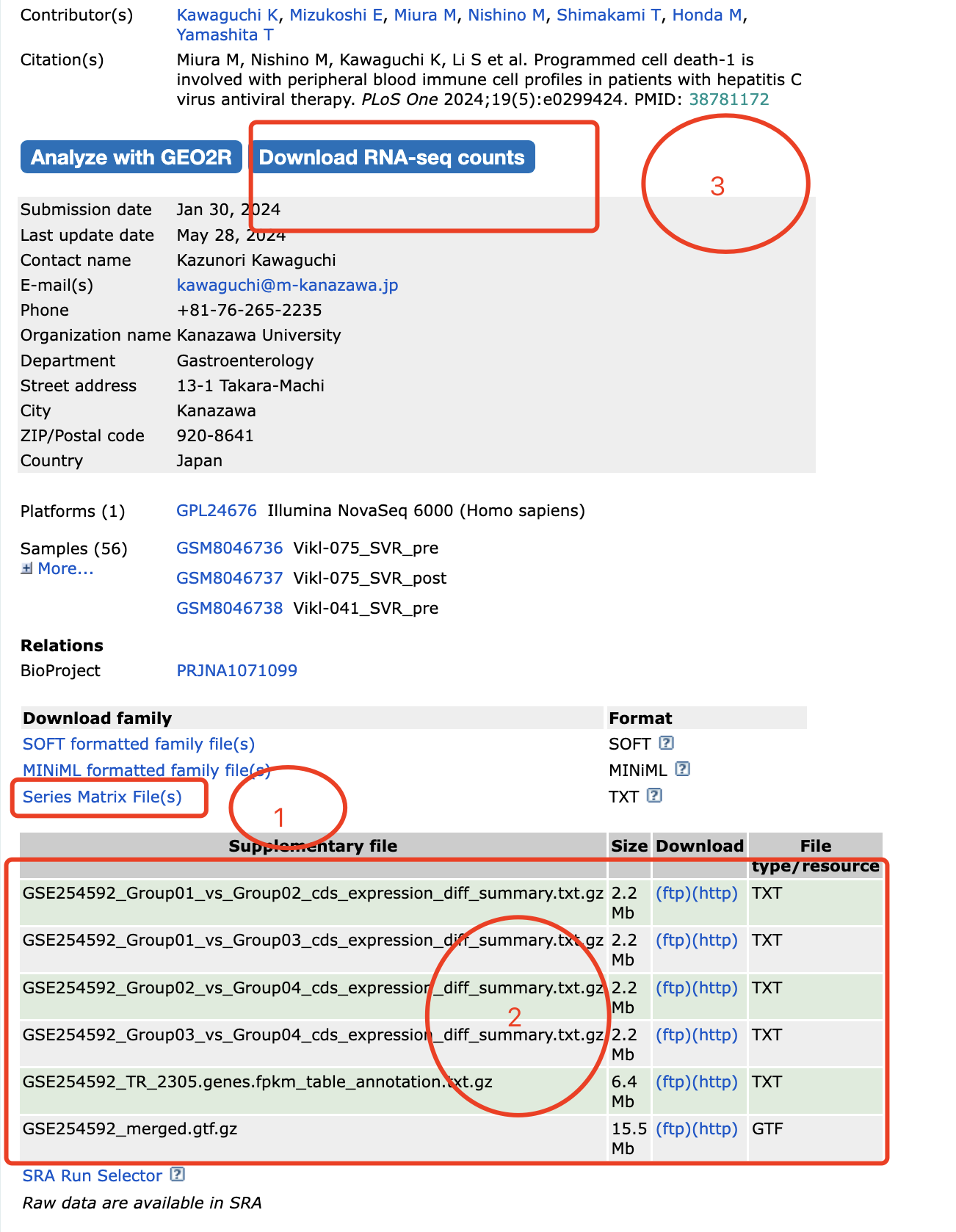
点进去就是geo官方的转录组定量流程后得到的表达量矩阵文件 ,如下所示链接 :
- https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE254592
- https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&acc=GSE254592&format=file&file=GSE254592_raw_counts_GRCh38.p13_NCBI.tsv.gz
而且是每个数据集都是同样的结构
任意数据集,下面的URL只需要替换里面的gse编号即可:
- https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122709
- https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE122709
每个数据集都有自己的表达量矩阵文件下载,要么是作者自己给出来的转录组测序定量好的矩阵,要么是geo官方统一定量的文件,如下所示:
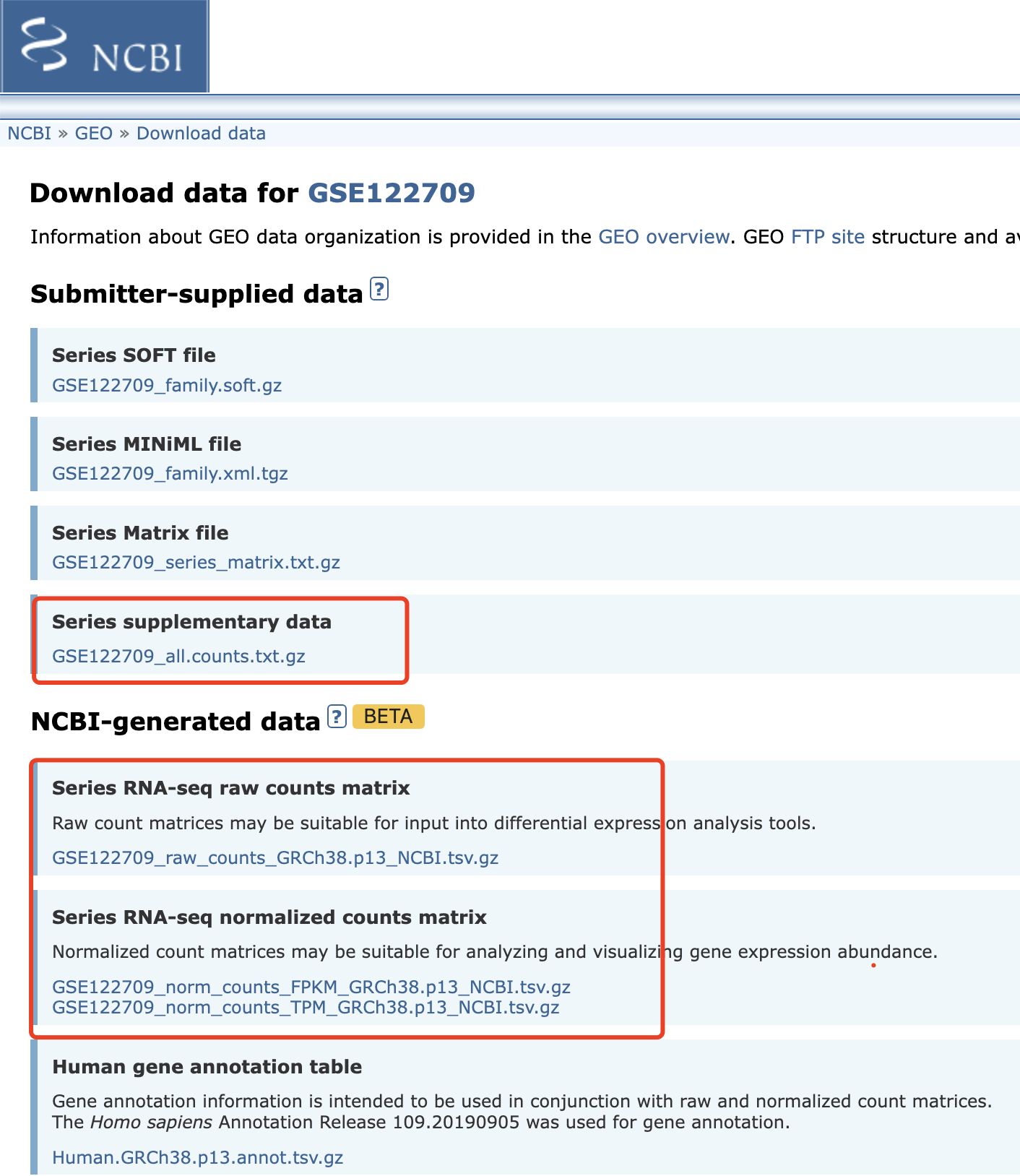
也可以看它们的URL的规则:
- https://ftp.ncbi.nlm.nih.gov/geo/series/GSE122nnn/GSE122709/suppl/GSE122709_all.counts.txt.gz
- https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&acc=GSE122709&format=file&file=GSE122709_raw_counts_GRCh38.p13_NCBI.tsv.gz
实战一下
让我们看看2024年5月31日,中山大学附属孙逸仙纪念医院苏士成、陆艺文等人在癌症领域顶级期刊 Cancer Cell 上发表了题为:Tumor cells impair immunological synapse formation via central nervous system-enriched metabolite 的研究论文,该研究里面有一个公开的单细胞转录组数据集,以及转录组测序数据集:
可以看到,这个转录组测序是9个样品,分成两组:
GSM7429237 resistant1
GSM7429238 resistant2
GSM7429239 resistant3
GSM7429240 resistant4
GSM7429241 sensitive1
GSM7429242 sensitive2
GSM7429243 sensitive3
GSM7429244 sensitive4
GSM7429245 sensitive5
文章里面也给出来了转录组差异分析结果:

但是如果大家直接从geo界面看是每个样品的fpkm矩阵 :
GSM7429237_resistant1_pt01002_S19187.txt.gz 915.7 Kb
GSM7429238_resistant2_pt01003_S20199.txt.gz 923.3 Kb
GSM7429239_resistant3_pt01004_S20223.txt.gz 928.3 Kb
GSM7429240_resistant4_pt01009_S1820.txt.gz 923.1 Kb
GSM7429241_sensitive1_pt01006_S2040.txt.gz 903.4 Kb
GSM7429242_sensitive2_pt01007_S20177.txt.gz 911.0 Kb
GSM7429243_sensitive3_pt01010_S20195.txt.gz 922.3 Kb
GSM7429244_sensitive4_pt01011_S20191.txt.gz 907.0 Kb
GSM7429245_sensitive5_pt01012_S2017.txt.gz 919.7 Kb
就不适合做差异分析,所以可以下载这个 GSE233484_raw_counts_GRCh38.p13_NCBI.tsv.gz 文件 然后读取:
# load counts table from GEO
urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?format=file&type=rnaseq_counts"
path <- paste(urld, "acc=GSE233484", "file=GSE233484_raw_counts_GRCh38.p13_NCBI.tsv.gz", sep="&");
path='GSE233484_raw_counts_GRCh38.p13_NCBI.tsv.gz'
tbl <- as.matrix(data.table::fread(path, header=T, colClasses="integer"), rownames=1)
简单的做了常规的转录组测序 差异分析,基本上跟上面的题为:Tumor cells impair immunological synapse formation via central nervous system-enriched metabolite 的研究论文里面的图表一致,说明文章的数据分析是准确无误的!