安排学徒复现一个新鲜出炉的阿兹海默症的单细胞文章:《Characterisation of premature cell senescence in Alzheimer’s disease using single nuclear transcriptomics》:
- Acta Neuropathologica (2024)
- https://doi.org/10.1007/s00401-024-02727-9
对应的数据集是:GSE264648,大家很容易读取作者提供的表达量矩阵然后进行降维聚类分群,但是学徒在进行 the ‘canonical senescence pathway (CSP)’, the ‘senescence initiating pathway (SIP)’ 的打分的时候,虽然是复现出来了文章的主要是结论, 就是: - the aver- age expression of individual senescence gene was most in microglia (e.g., ATM, RB1, NFATC2 and GLB1)
如下所示被高亮的区域就是小胶质细胞,确实是打分会比较高,但是不够显著。所以我让它简单的询问了人工智能大模型的解决方案。做了一个简单的配色, 就可以达到显著的效果,代码是:my.colors <- colorRampPalette(c("lightblue", "white", "darkred"))(100) FeaturePlot(sc_dataset,'gene_vector_UCell' ) + FeaturePlot(sc_dataset,'gene_vector_UCell',cols = my.colors)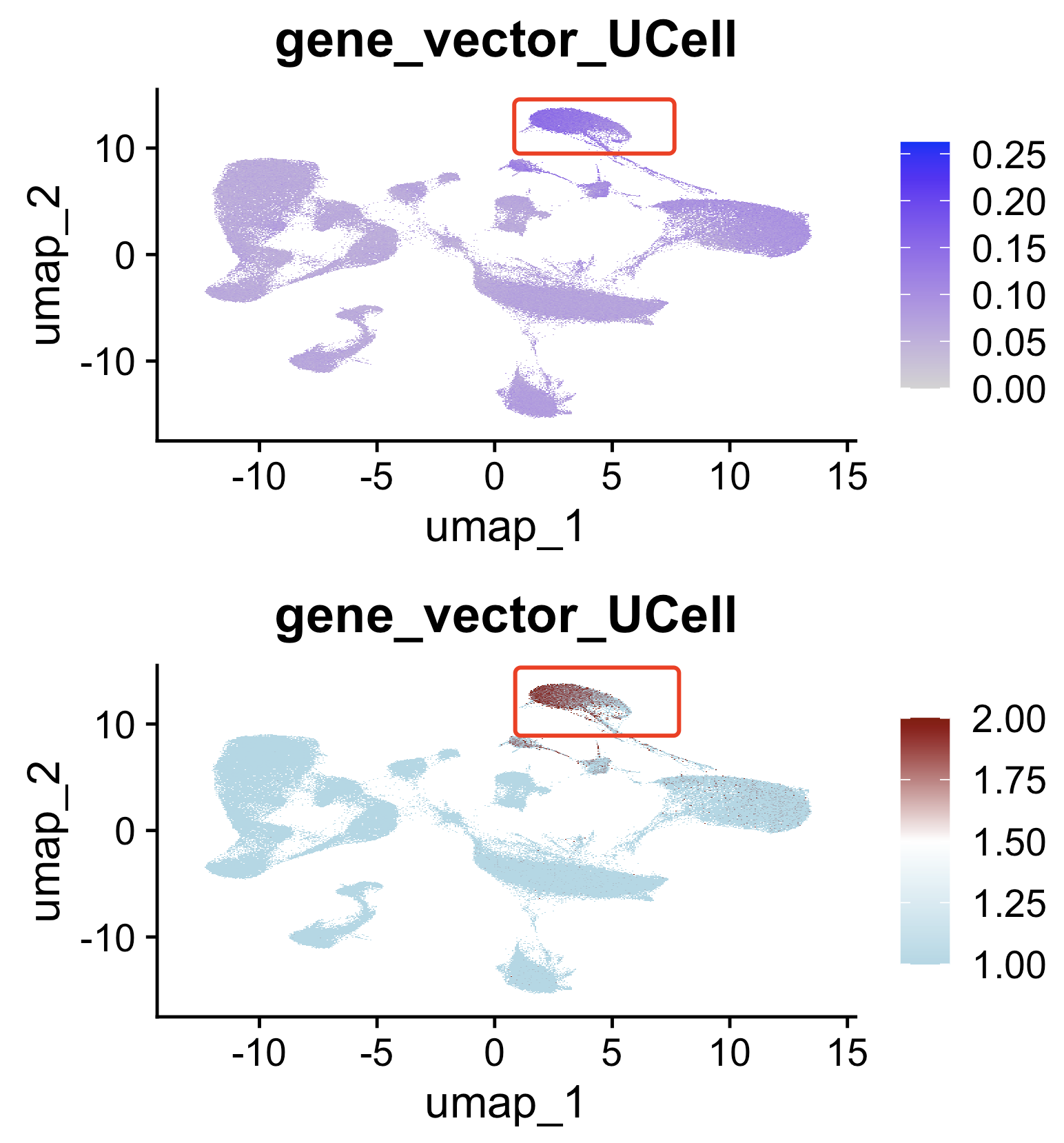
提问:r编程语言里面的Seurat包里面的FeaturePlot函数可视化一个基因的时候颜色渐变不够明显,有什么调色方案吗
在R语言的Seurat包中,FeaturePlot函数用于可视化单个或多个基因在各个细胞中的表达水平。如果颜色渐变不够明显,可以通过调整颜色渐变的参数来增强视觉效果。以下是一些常用的调色方案和参数调整的建议:
- 使用已有的颜色渐变方案:
Seurat内置了一些颜色渐变方案,例如”viridis”、”cividis”、”magma”、”inferno”、”plasma”等,这些颜色方案通常提供较好的对比度和渐变效果。 - 自定义颜色渐变:
你可以使用R的颜色函数,如colorRampPalette()或brewer.pal()来自定义颜色渐变。例如:my.colors <- colorRampPalette(c("blue", "white", "red"))(100)这段代码创建了一个从蓝色到白色再到红色的颜色渐变。
- 调整颜色的数量:
通过调整颜色的数量,可以改变渐变的细腻程度。颜色数量越多,渐变越细腻。 - 调整底图的分辨率:
使用FeaturePlot函数时,可以通过nCol参数调整底图的列数,以及pt.size参数调整点的大小,从而影响视觉效果。 - 使用
reorder参数:
使用reorder参数可以根据基因表达量的大小重新排序细胞,可能会使表达模式更加明显。 - 调整透明度:
通过设置alpha参数,可以调整点的透明度,有助于在细胞点重叠时区分不同的表达水平。 - 使用对数转换:
如果基因表达水平差异很大,使用对数转换可能会使渐变更加明显。可以通过Log1p函数对数据进行转换。 - 使用
Scale参数:
FeaturePlot函数的scale参数可以调整颜色的缩放,例如使用scale = "count"根据基因计数缩放颜色。
下面是一个使用自定义颜色渐变的FeaturePlot示例:library(Seurat) library(ggplot2) # 假设seurat_obj是你的Seurat对象,gene是你想可视化的基因 my.colors <- colorRampPalette(c("lightblue", "white", "darkred"))(100) FeaturePlot(seurat_obj, features = gene, cols = my.colors)请注意,这些调整可能需要根据你的具体数据和可视化需求进行尝试和优化。如果需要更专业的调色方案,可以考虑使用如
ggplot2的scale_color_gradientn()函数来获取更多的颜色选项。但是为什么修改配色的同时造成了值的修改
如果仔细看上面的两个FeaturePlot就会发现最开始默认配色的时候打分在0到0.25这个范围,但是配色后值的范围成为了0到1,这个是为什么呢?