安排学徒复现一个新鲜出炉的阿兹海默症的单细胞文章:《Characterisation of premature cell senescence in Alzheimer’s disease using single nuclear transcriptomics》:
- Acta Neuropathologica (2024)
- https://doi.org/10.1007/s00401-024-02727-9
对应的数据集是:GSE264648,大家很容易读取作者提供的表达量矩阵然后进行降维聚类分群,如下所示的7个亚群泾渭分明,干净的让人神往!
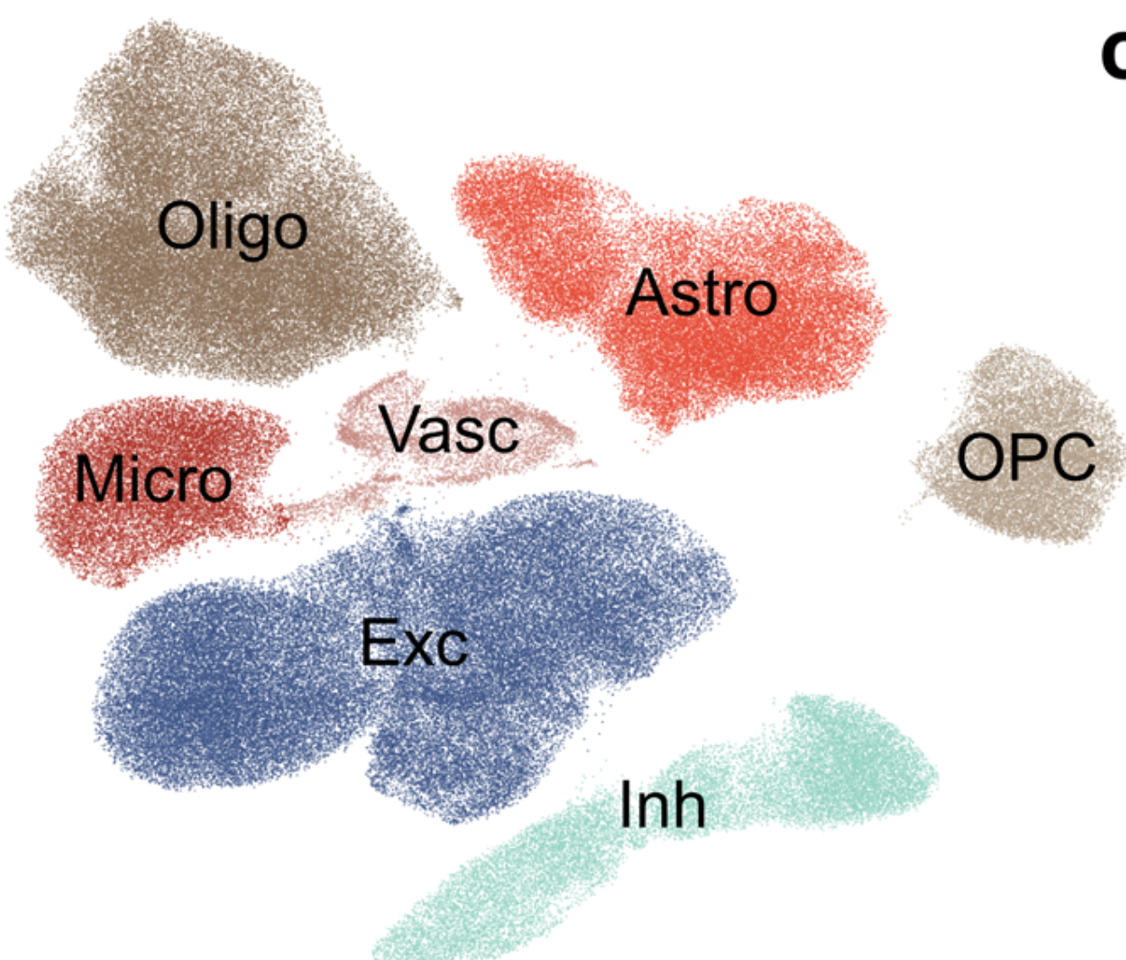
我们可以读取作者提供的文件:
ct=readRDS('inputs/GSE264648_counts_all_celltype.rds')
ct
meta= readRDS('inputs/GSE264648_metadata_all_celltype.rds')
head(meta)
identical(colnames(ct),rownames(meta))
sce.all=CreateSeuratObject(counts = ct ,
meta.data = meta)
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
sce.all
但是降维聚类分群结果跟文章差异就很大,如下所示:
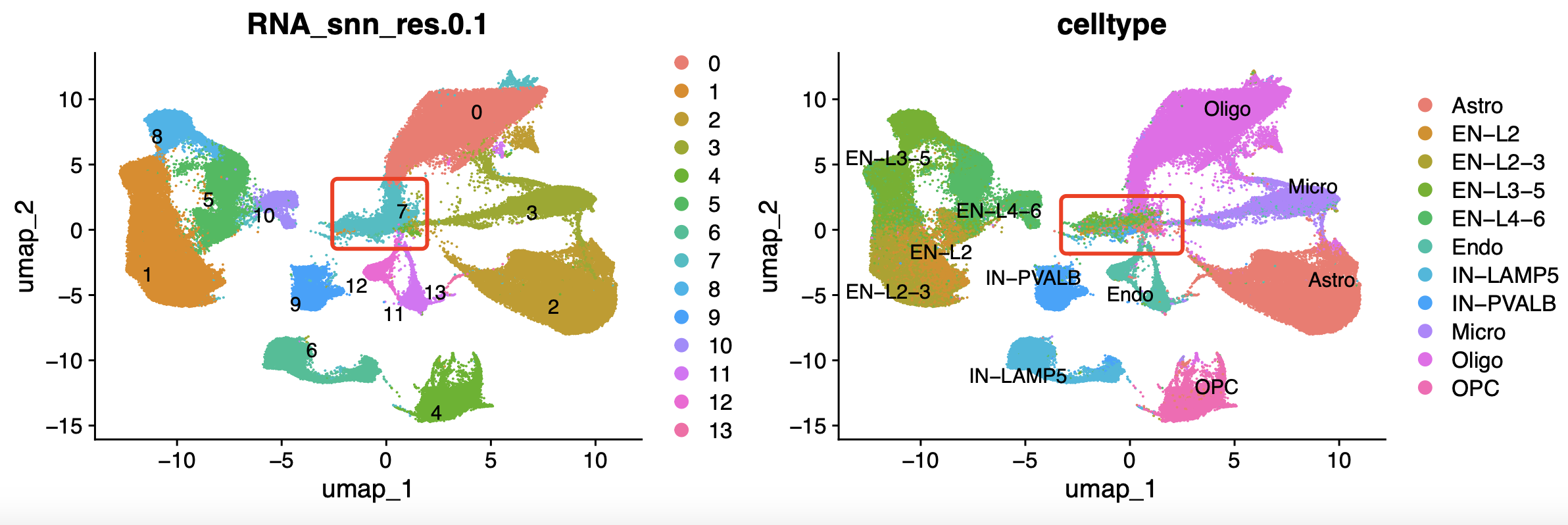
可以看到,我们有一个7亚群,是质量很低的,其实是可以提高过滤的标准:
sce.all.filt
sce.all.filt=subset(sce.all.filt,subset = percent_mito < 20 &
# nCount_RNA <10000 &
# nFeature_RNA <7500 &
nFeature_RNA>500)
这样的话,可以删除1万左右的细胞,剩下的19万细胞的降维聚类分群结果如下所示:
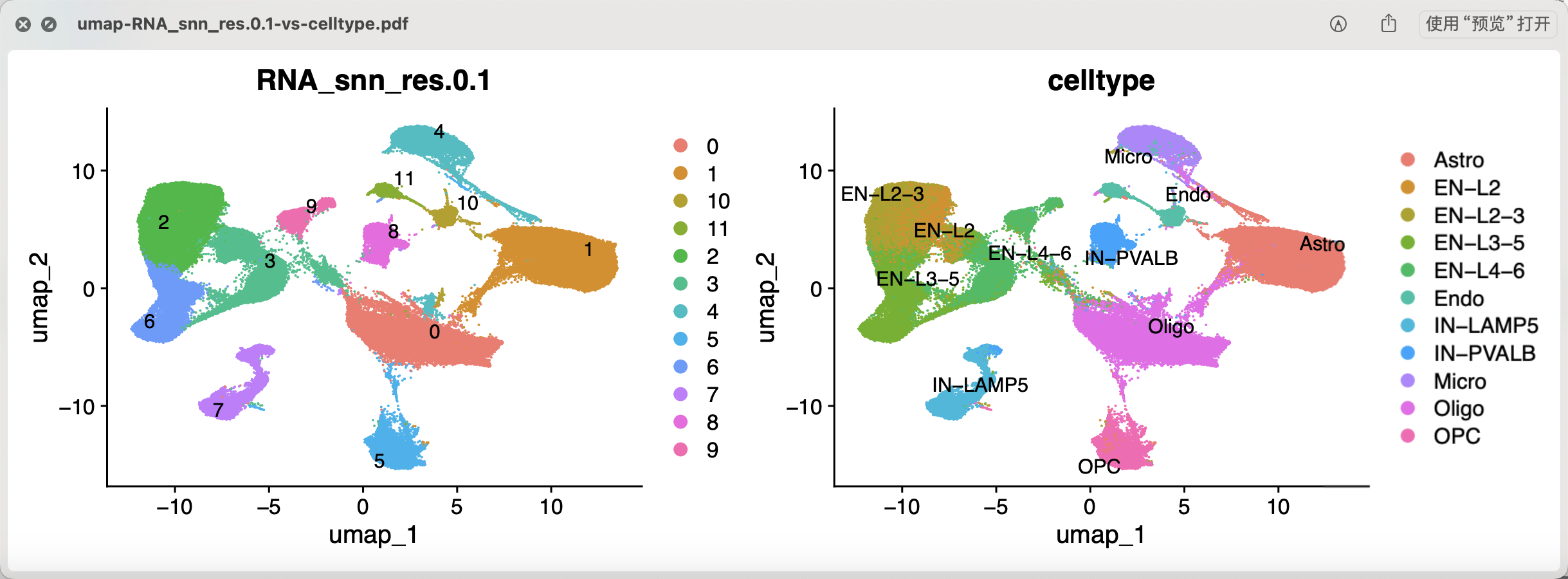
如果我们仔细看作者提供的信息,其实是有 Liger 这个算法做多样品整合,而我们通常是harmony:
2.9M Aug 21 19:18 GSE264648_UMAP_Liger_all_celltype.rds
1.4G Aug 21 19:18 GSE264648_counts_all_celltype.rds
8.4M Aug 21 19:18 GSE264648_metadata_all_celltype.rds
我让大模型帮我整理了一下Liger和Harmony是两种用于多样本单细胞转录组数据整合的算法:
它们各自有不同的特点、优势和局限性 :
Liger (Likelihood-based Integration of Genomic Datasets with Replication)
特点:
- Liger使用贝叶斯分层模型来整合数据集。
- 它考虑了技术变异性和生物学变异性。
- Liger适用于整合具有或没有重复的转录组数据集。
优点:
- 能够处理不同平台和批次的数据集。
- 通过考虑重复,可以更准确地估计变异性。
- 灵活处理缺失数据。
局限性:
- 计算成本可能较高,特别是在处理大型数据集时。
- 需要对贝叶斯统计有一定的了解。
Harmony (Hierarchical Alignment and Orthogonal Modeling)
特点:
- Harmony使用线性回归模型来整合数据集。
- 它通过正交分解来去除批次效应。
- Harmony适用于大规模数据集的整合。
优点:
- 计算效率高,适合处理大规模数据集。
- 易于实现和使用。
- 可以通过调整参数来控制整合的严格程度。
局限性:
- 对于小样本数据集可能不够敏感。
- 在某些情况下可能无法完全去除批次效应。
异同点
相似之处:
- 两者都旨在整合来自不同批次或平台的单细胞转录组数据。
- 都试图去除批次效应,以揭示生物学变异性。
不同之处:
- 方法论:Liger基于贝叶斯统计,而Harmony基于线性回归和正交分解。
- 计算效率:Harmony通常更快,尤其是在大数据集上。
- 灵活性:Liger可能在处理不同类型数据时更加灵活。
- 统计基础:Liger需要用户对贝叶斯统计有更深的理解,而Harmony的方法更直观易懂。
应用场景
- 当数据集具有不同来源、平台,并且需要考虑重复时,Liger可能是更好的选择。
- 当处理大规模数据集,并且需要快速整合时,Harmony可能更合适。
结论
选择哪种算法取决于具体的数据特性、计算资源和用户对统计方法的熟悉程度。在实际应用中,可能需要尝试多种方法,以确定哪种方法最适合特定的数据集和研究目标。此外,数据预处理和后处理步骤也对最终的整合效果有重要影响。