众所周知,转录组测序后拿到的表达量矩阵通常是基因在样品的reads的数量,就是最原始的整数的counts矩阵啦。它有两个归一化方向,首先是样品方向的就是抹去各个样品的文库大小这个变量,然后是基因方向的就是抹去基因长度对表达量的影响。
如果是使用deseq2这样的包进行转录组测序的表达量的差异分析需要的是最原始的整数的counts矩阵即可,如果是做表达量热图,通常是使用归一化后的矩阵,可以是两个方向都做。如果仅仅是考虑文库大小就是cpm和rpm,如果同时考虑基因长度就是 FPKM(Fragments Per Kilobase of transcript per Million mapped reads),以及tpm,让我们来理解一下:
cpm和rpm是同一个概念
CPM和RPM是同一种基因表达量标准化方法,它们用于调整测序深度的差异,以便在不同样本之间进行比较,英文全称是:
- CPM (Counts Per Million):
- RPM (Reads Per Million):
其实就是就是最原始的整数的counts矩阵除以每个样品的文库大小(以1M为单位),但是目前转录组测序非常标准化了其实文库大小统一是20M附近,如果不做这个cpm或者rpm,问题也不大,但是就怕碰到极端值情况。
tpm不一定是转录本定量
本来呢,应该是先理解 FPKM(Fragments Per Kilobase of transcript per Million mapped reads),就是上面的cpm或者rpm矩阵再除以每个基因的长度(以1kb为单位)情况。但是这样的FPKM表达量有一个弊端就是每个样品的所有的基因的FPKM加和并不是固定的,所以就引入了tpm概念,就是继续除以FPKM表达量的文库(以1M为单位)大小,这个时候就不一定是20M附近,因为每个样品的FPKM加和并不是固定的。但是TPM(Transcripts Per Million)看起来很容易让人误解是针对转录本的定量。
最原始的整数的counts矩阵的差异分析
只需要在你的r里面加载两个包,就可以完成下面的分析啦:
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
# BiocManager::install('airway')
# 加载airway数据集并转换为表达矩阵
library(airway,quietly = T)
data(airway)
rawcount <- assay(airway)
group_list <- colData(airway)$dex
# 过滤在至少在75%的样本中都有表达的基因 (可选步骤,也可以修改)
keep <- rowSums(rawcount>0) >= floor(0.75*ncol(rawcount))
table(keep)
filter_count <- rawcount[keep,]
filter_count[1:4,1:4]
dim(filter_count)
run_deseq2 <- function(exprSet,group_list){
library(DESeq2)
# 第一步,构建DESeq2的DESeq对象
colData <- data.frame(row.names=colnames(exprSet),group_list=group_list)
dds <- DESeqDataSetFromMatrix(countData = exprSet,colData = colData,design = ~ group_list)
# 第二步,进行差异表达分析
dds2 <- DESeq(dds)
# 提取差异分析结果,trt组对untrt组的差异分析结果
tmp <- results(dds2,contrast=c("group_list","trt","untrt"))
DEG_DESeq2 <- as.data.frame(tmp[order(tmp$padj),])
head(DEG_DESeq2)
# 去除差异分析结果中包含NA值的行
DEG_DESeq2 = na.omit(DEG_DESeq2)
}
deg_raw = run_deseq2(filter_count,group_list)
上面的代码里面,我定义了一个 run_deseq2 函数,方便后续调用:
针对cpm或者rpm矩阵的差异分析
假如极端情况下,你拿到了的转录组测序的表达量矩阵就是cpm或者rpm,你可以直接把矩阵乘以20后向上取整,如下所示的代码:
ct2 = floor(20*edgeR::cpm(filter_count))
deg_cpm = run_deseq2(ct2,group_list)
save(deg_raw,deg_cpm,file = 'deg.Rdata')
可以看到之前的整数的counts矩阵里面每个样品的文库大小确实是不一样的,但是都是在20M附近,而如果你拿到了的转录组测序的表达量矩阵就是cpm或者rpm意味着你没办法知道每个样品的真实文库大小,因为被抹除了 。直接把矩阵乘以20后向上取整的后果就是每个样品很整齐,就是20M的文库大小;
> colSums(filter_count)/1e6
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
20.63292 18.80417 25.34134 15.16004 24.44175 30.81030
SRR1039520 SRR1039521
19.11741 21.15675
> colSums(ct2)/1e6
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
19.98724 19.99079 19.98892 19.98928 19.98938 19.98891
SRR1039520 SRR1039521
19.99101 19.98787
比较两次差异分析结果:
两次都是同样的 run_deseq2 函数,所以结果矩阵的格式是一致的:
rm(list = ls())
library(data.table)
load('deg.Rdata')
ids=intersect(rownames(deg_cpm),
rownames(deg_raw))
df= data.frame(
deg_cpm = deg_cpm[ids,'log2FoldChange'],
deg_raw = deg_raw[ids,'log2FoldChange']
)
library(ggpubr)
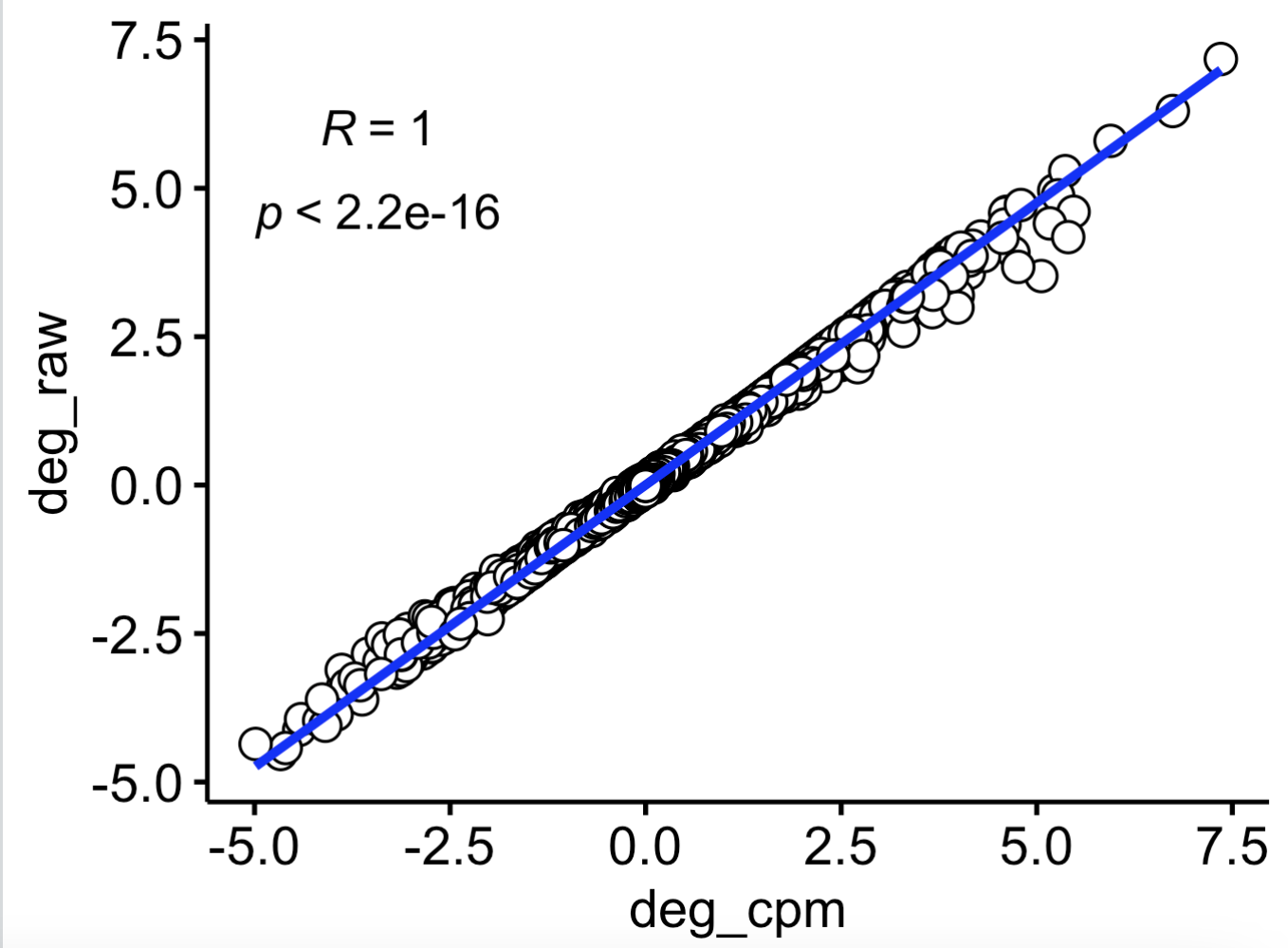
ggscatter(df, x = "deg_cpm", y = "deg_raw",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
可以看到虽然是两次计算的logFC略微有差异,但是相关性几乎是完美的:
也可以看看,两次差异分析后的统计学显著的上下调基因的一致性情况,代码如下所示:
modify_deg<-function(DEG_DESeq2){
# 筛选上下调,设定阈值
fc_cutoff <- 1
fdr <- 0.05
DEG_DESeq2$regulated <- "normal"
loc_up <- intersect(which(DEG_DESeq2$log2FoldChange>log2(fc_cutoff)),
which(DEG_DESeq2$padj<fdr))
loc_down <- intersect(which(DEG_DESeq2$log2FoldChange< (-log2(fc_cutoff))),
which(DEG_DESeq2$padj<fdr))
DEG_DESeq2$regulated[loc_up] <- "up"
DEG_DESeq2$regulated[loc_down] <- "down"
table(DEG_DESeq2$regulated)
head(DEG_DESeq2)
library(AnnoProbe)
ag=annoGene(rownames(DEG_DESeq2),
ID_type = 'ENSEMBL',species = 'human'
)
head(ag)
DEG_DESeq2$ENSEMBL=rownames(DEG_DESeq2)
deg_anno=merge(ag,DEG_DESeq2,by='ENSEMBL')
deg_anno=deg_anno[!duplicated(deg_anno$SYMBOL),]
rownames(deg_anno)=deg_anno$SYMBOL
return(deg_anno)
}
deg_cpm=modify_deg(deg_cpm)
deg_raw=modify_deg(deg_raw)
colnames(deg_cpm)
ids=intersect(rownames(deg_cpm),
rownames(deg_raw))
df= data.frame(
deg_cpm = deg_cpm[ids,'regulated'],
deg_raw = deg_raw[ids,'regulated']
)
table(df)
gplots::balloonplot(table(df))
可以看到的是两次的差异分析误差几乎是可以忽略不计的 :
> table(df)
deg_raw
deg_cpm down normal up
down 1111 14 0
normal 32 14683 13
up 0 8 1511
也可以进一步的看看两次差异分析的冲突的基因列表的功能情况:
symbols_list = split(ids,paste(df[,1],df[,2]))
library(clusterProfiler)
library(org.Hs.eg.db)
library(ReactomePA)
library(ggplot2)
library(stringr)
# 首先全部的symbol 需要转为 entrezID
gcSample = lapply(symbols_list, function(y){
y=as.character(na.omit(AnnotationDbi::select(org.Hs.eg.db,
keys = y,
columns = 'ENTREZID',
keytype = 'SYMBOL')[,2])
)
y
})
gcSample
pro='test'
# 第1个注释是 KEGG
xx <- compareCluster(gcSample, fun="enrichKEGG",
organism="hsa", pvalueCutoff=0.3)
dotplot(xx) + theme(axis.text.x=element_text(angle=45,hjust = 1)) +
scale_y_discrete(labels=function(x) str_wrap(x, width=50))
ggsave(paste0(pro,'_kegg.pdf'),width = 10,height = 8)
蛮有意思的, 这些基因都是代谢相关的:
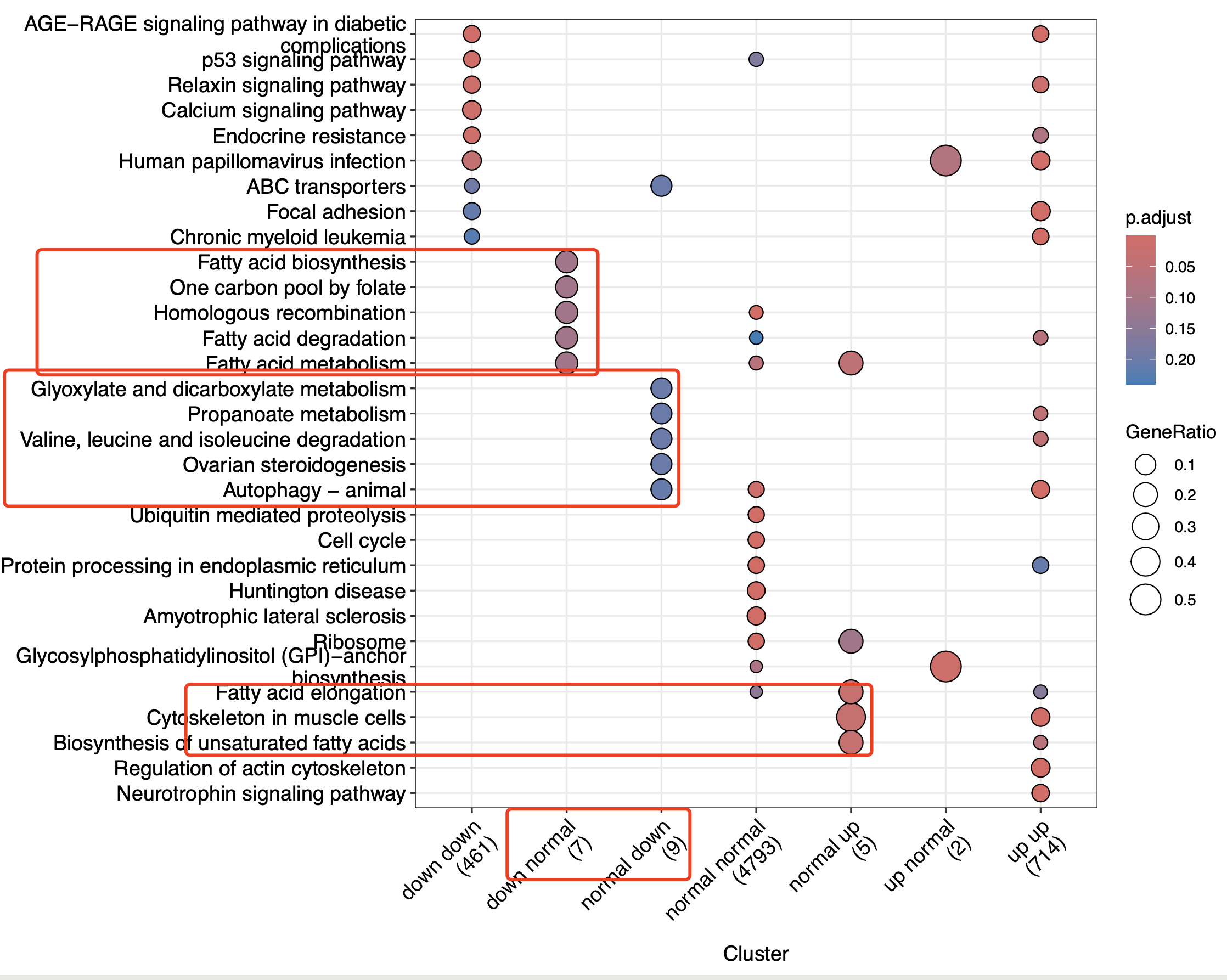
其实是可以深入探索一下:
$`down normal`
[1] "HDAC9" "NFYC" "NBN" "ALMS1" "MTHFD1L" "ACSBG2"
[7] "YIPF2" "PI4KB" "LIMS1" "INSIG1" "ZSCAN23" "PLPP4"
[13] "SP5" "GUSBP16"
$`normal down`
[1] "ABCB5" "CDH10" "SLC6A16" "PTGS2" "MOK"
[6] "PITPNM3" "C14orf93" "ISYNA1" "TBC1D30" "MCEE"
[11] "ZNF436" "RSAD2" "MDH1B" "DEPTOR" "LRRC56"
[16] "STOX2" "PODN" "RIN1" "GUSBP1" "CDNF"
[21] "SAMD11" "ZNF682" "FANK1" "EIF2AK3-DT" "H3P6"
[26] "AC108488.1" "ZNF512" "ZNF286B" "KLRA1P" "AL355102.1"
为什么看起来基本上没有修改表达量矩阵的操作,也会有一些冲突的基因呢?作为学徒作业给大家吧!
下一期我们说一下如果你的矩阵被fpkm了或者tpm了,该如何最佳差异分析呢?