下载地址:http://hgdownload.cse.ucsc.edu/admin/exe/
我一般是使用linux版本的:wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/liftOver
使用方法:【从hg38转到hg19】
因为主流的基因组版本还是hg19,但是时代在进步,已经有很多信息都是以hg38的形式公布出来的了。
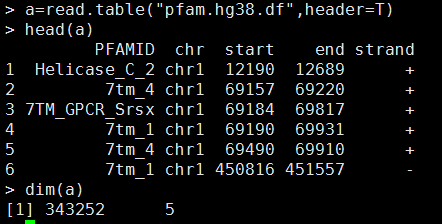
比如,我下载了pfam.df这个protein domain注释文件,对人的hg38基因组每个坐标都做了domain注释,数据形式如下:
查看文件内容head pfam.hg38.df ,如下:
PFAMID chr start end strand
Helicase_C_2 chr1 12190 12689 +
7tm_4 chr1 69157 69220 +
7TM_GPCR_Srsx chr1 69184 69817 +
7tm_1 chr1 69190 69931 +
7tm_4 chr1 69490 69910 +
7tm_1 chr1 450816 451557 -
7tm_4 chr1 450837 451263 -
EPV_E5 chr1 450924 450936 -
7TM_GPCR_Srsx chr1 450927 451572 -
我想把domain的起始终止坐标转换成hg19的,就必须要借助UCSC的liftover这个工具啦
而且它只能对bed等符合要求的格式进行转换
示例如下:
chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0 chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
很简单的,把自己的文件随便凑几列信息,做成这个9列的格式即可
cat pfam.hg38.df |sed 's/\r//g' |awk '{print $2,$3,$4,$1,0,$5,$3,$4,"255,0,0"}' >pfam.hg38.bed
这样就有了足够的文件可以进行坐标转换啦,转换的命令非常简单!
chmod 777 liftOver
./liftOver pfam.hg38.bed hg38ToHg19.over.chain pfam.hg19.bed unmap
然后运行成功了会有 提示,报错一般是你的格式不符合标准bed格式,自己删掉注释行等等不符合的信息即可
Reading liftover chains
Mapping coordinates
转换后,稍微检查一下就可以看到坐标的确发生了变化,当然,我们只需要看前面几列信息即可
grep -w p53 *bed
pfam.hg19.bed:chr11 44956439 44959858 p53-inducible11 0 - 44956439 44959858 255,0,0
pfam.hg19.bed:chr11 44956439 44959767 p53-inducible11 0 - 44956439 44959767 255,0,0
pfam.hg19.bed:chr2 669635 675557 p53-inducible11 0 - 669635 675557 255,0,0
pfam.hg19.bed:chr22 35660826 35660982 p53-inducible11 0 + 35660826 35660982 255,0,0
仔细看看坐标是不是变化啦!
pfam.hg38.bed:chr11 44934888 44938307 p53-inducible11 0 - 44934888 44938307 255,0,0
pfam.hg38.bed:chr11 44934888 44938216 p53-inducible11 0 - 44934888 44938216 255,0,0
pfam.hg38.bed:chr2 669635 675557 p53-inducible11 0 - 669635 675557 255,0,0
pfam.hg38.bed:chr22 35264833 35264989 p53-inducible11 0 + 35264833 35264989 255,0,0
其实R里面的bioconductor系列包也可以进行坐标转换 http://www.bioconductor.org/help/workflows/liftOver/
这个可以直接接着下载pfam.df数据库来做下去。更方便一点。
我的数据如下,需要自己创建成一个GRanges对象
library(GenomicRanges)
pfam.hg38 <- GRanges(seqnames=Rle(a[,2]),
ranges=IRanges(a[,3], a[,4]),
strand=a[,5])
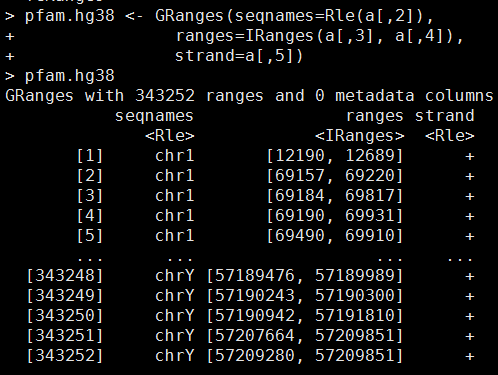
这样就OK拉,虽然这只是一个很简陋的GRanges对象,但是这个GRanges对象可以通过R的liftover方法来转换坐标啦。
library(rtracklayer)
ch = import.chain("hg38ToHg19.over.chain")
pfam.hg19 = liftOver(pfam.hg38, ch)
pfam.hg19 =unlist(pfam.hg19)
再把这个转换好的pfam.hg19 写出即可