表达芯片大家最熟悉的当然是affymetrix系列芯片啦,而且分析套路很简单,直接用R的affy包,就可以把cel文件经过RMA或者MAS5方法得到表达矩阵。illumina出厂的芯片略微有点不一样,它的原始数据有3个层级,一般拿到的是Processed data (示例), 当仍然需要一系列的统计学方法才能提取到表达矩阵。我比较喜欢用bioconductor,所以下面讲一讲如何用lumi包来处理这个芯片数据!
这个lumi包的使用代码和说明书都有,按部就班的学一遍就好了。
如果仅仅是分析数据,那么并不难,但是每个分析步骤后面都隐含着一系列的统计学方法,想彻底搞清楚他它们, 就很难了。
data(example.lumi)
lumi.N.Q <- lumiExpresso(example.lumi)
dataMatrix <- exprs(lumi.N.Q)
重点就是得到表达矩阵,它封装好了一个函数,lumiExpresso可以直接处理LumiBatch对象,这个函数结合了,N,T,B,Q(normalization,transformation,backgroud correction,qulity control)四个步骤,其中Q这个步骤又包括8种统计学图片。在该包的文章有详细说明:http://bioinformatics.oxfordjournals.org/content/24/13/1547.full
而 LumiBatch 对象是通过 lumiR.batch 读取的芯片文件被Illumina Bead Studio toolkit 处理的结果,也就是通常我们从公司或者GEO下载的数据( level 3 的 process data),如下所示:
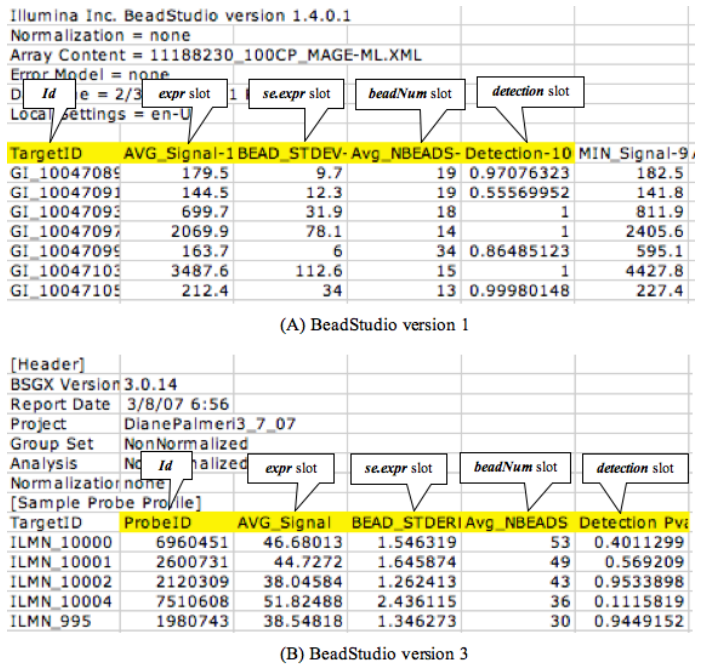
这个包用的测试文件Barnes_gene_profile.txt可以在http://www.chibi.ubc.ca/wp-content/uploads/2013/02/ 下载。
如果是在GEO下载公共数据,每个study都会给芯片描述文件,基本没有用,只需要下载non-normalized.txt.gz类似的文件就好了
GPL10558_HumanHT-12_V4_0_R1_15002873_B.txt.gz 13.1 Mb
GPL10558_HumanHT-12_V4_0_R2_15002873_B.txt.gz 13.1 Mb
比如我下载了:ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE30nnn/GSE30669/suppl/GSE30669_HEK_Sample_Probe_Profile.txt.gz 这个文件,就可以直接用lumi包的lumiR.batch 函数读取文件成为LumiBatch对象,然后被lumiExpresso函数直接处理,然后被exprs函数提取表达矩阵。
rm(list=ls())library(lumi)# setwd('G:/array/illumina-beadseed-v4/lumi_example')# fileName <- 'Barnes_gene_profile.txt' # Not Run## 首先是从illumina的芯片结果文件,自己用R的lumi包来获取表达矩阵。setwd('G:/array/illumina-beadseed-v4/GSE30669')fileName <- 'GSE30669_HEK_Sample_Probe_Profile.txt' # Not Runx.lumi <- lumiR.batch(fileName) ##, sampleInfoFile='sampleInfo.txt')pData(phenoData(x.lumi))## Do all the default preprocessing in one steplumi.N.Q <- lumiExpresso(x.lumi)### retrieve normalized datadataMatrix <- exprs(lumi.N.Q)## 下面是从GEO里面下载表达矩阵rm(list=ls())library(GEOquery)library(limma)GSE30669 <- getGEO('GSE30669', destdir=".",getGPL = F)exprSet=exprs(GSE30669[[1]])GSE30669[[1]]pdata=pData(GSE30669[[1]])exprSet=exprs(GSE30669[[1]])很明显可以看到前面得到的dataMatrix 和后面得到的 exprSet 都是我们想要的表达矩阵
## 因为你有时候获取别人处理好的表达矩阵,不符合你的normalization要求。
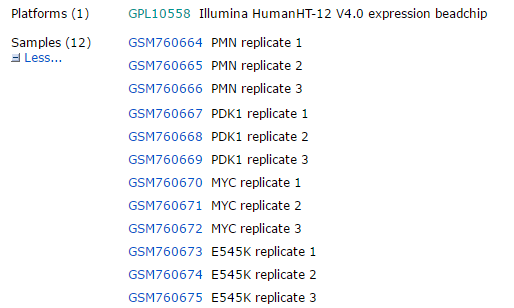
这个芯片一般是处理12个样本,从GEO里面很容易看到样品是如何分组的。
lumi这个包甚至还提供了一个函数produceGEOSubmissionFile来直接把我们的芯片数据转换成NCBI的GEO要求的格式
最后,官网链接很重要:https://support.illumina.com/array/array_kits/humanht-12_v4_expression_beadchip_kit/downloads.html