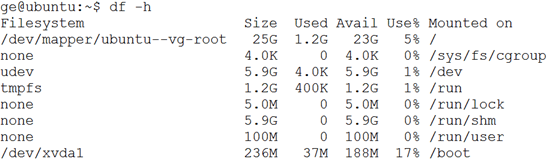
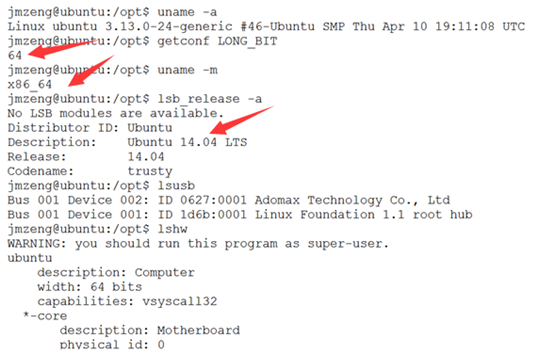
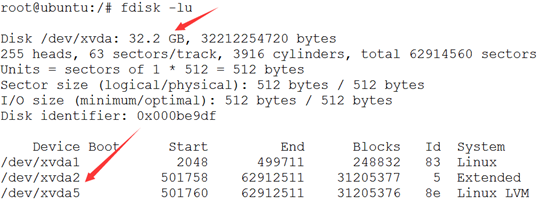
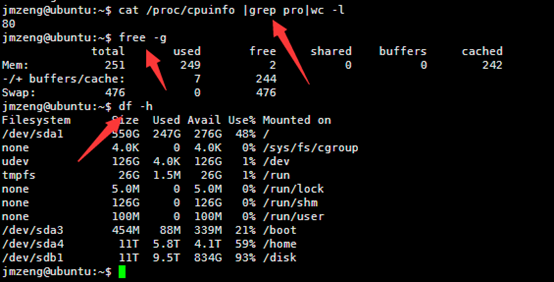
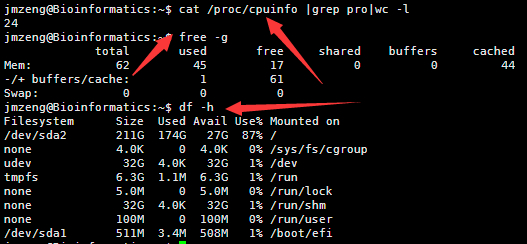
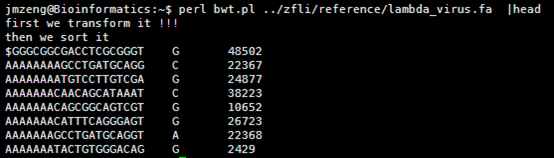
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
1、JDK官网上http://www.oracle.com/technetwork/java/javase/downloads/index.html选择:
但是,如果你的服务器是64位的,请不要选择i586,选择你自己的机器对应的!
2、将打开终端,建立目录:
Sudo mkdir /usr/lib/java
3、将下载的 jdk-7u3-linux-i586.tar.gz移到这个文件夹下面并进行解压,改名字:
sudo mv jdk-7u3-linux-i586.tar.gz /usr/lib/java
sudo tar –xvf jdk-7u3-linux-i586.tar.gz
mv jdk1.7.0_03java-7-sun
4、修改环境变量:
在终端输入:vim /etc/profile
然后添加以下代码:
export JAVA_HOME=/usr/lib/java/jdk1.8.0_25
export JRE_HOME=${JAVA_HOME}/jre
export CLASSPATH=.:${JAVA_HOME}/lib:${JRE_HOME}/lib
export PATH=${JAVA_HOME}/bin:$PATH
保存之后,再运行下面命令更新电脑的配置文件
source /etc/profile 这个千万要记得!!!!
5、在终端中输入 java –version,显示:
jeydragon@jeydragon-VirtualBox:~$ java -version
java version "1.7.0_03"
Java(TM) SE Runtime Environment (build 1.7.0_03-b04)
Java HotSpot(TM) Client VM (build 22.1-b02, mixed mode)
表示安装成功