发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
很多人可能并没有自己的服务器,那么就只能通过虚拟机来试试ubuntu啦
我想起来我以前玩虚拟机的时候遇到过一些困难,记录了一些,分享给大家, Continue reading
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
很多人可能并没有自己的服务器,那么就只能通过虚拟机来试试ubuntu啦
我想起来我以前玩虚拟机的时候遇到过一些困难,记录了一些,分享给大家, Continue reading
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
很多时候大家的服务器可能并不是想联网,只是想玩一下,或者只是因为生信的某些软件要求数据库,所以大家可能会单独安装mysql,或者想学习perl的CGI模块,需要apache。
非常简单只需要几条命令就可以完成。
1. sudo apt-get install mysql-server
2. sudo apt-get install mysql-client
3. sudo apt-get install libmysqlclient-dev
安装过程中会提示设置密码什么的,注意设置了不要忘了,安装完成之后可以使用如下命令来检查是否安装成功:
sudo netstat -tap | grep mysql
通过上述命令检查之后,如果看到有mysql 的socket处于 listen 状态则表示安装成功。
登陆mysql数据库可以通过如下命令:
mysql -u root -p
-u 表示选择登陆的用户名, -p 表示登陆的用户密码,上面命令输入之后会提示输入密码,此时输入密码就可以登录到mysql。
在Ubuntu上安装Apache,有两种方式:1 使用开发包的打包服务,例如使用apt-get命令;2 从源码构建Apache。本文章将详细描述这两种不同的安装方式。
方法一:使用开发包的打包服务——apt-get
安装apache,在命令行终端中输入一下命令:
$ sudo apt-get install apache2
安装完成后,重启apache服务,在命令行终端中输入一下命令:
$ sudo /etc/init.d/apache2 restart
可能会出现的问题1: NameVirtualHost *:80 has no VirtualHosts,
出现上述问题的原因:定义了多个NameVirtualHost,故将/etc/apache2/ports.conf中的NameVirtualHost *:80注释掉即可。
可能会出现的问题2: Could not reliably determine the server's fully qualified domain name, using 127.0.1.1 for ServerName
原因:
根据提示,无法可靠的确定服务器的有效域名,使用127.0.1.1作为服务器域名。应此,在下面的测试中,应该使用127.0.1.1,而不是127.0.0.1!
解决:
$ vim /etc/apache2/httpd.conf,在文件中添加:
ServerName localhost:80,再次重启apache2,就可以使用127.0.0.1来访问web服务器啦!
测试:
在浏览器里输入http://localhost或者是http://127.0.0.1,如果看到了It works!,那就说明Apache就成功的安装了,Apache的默认安装,会在/var下建立一个名为www的目录,这个就是Web目录了,所有要能 过浏览器访问的Web文件都要放到这个目录里。
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
主流的网络服务器配置就是linux+apache+mysql+php咯,简称LAMP
在ubuntu系统里面安装这个是非常easy的
sudo apt-get install apache2 mysql-server mysql-client php5 php5-gd php5-mysql Continue reading
此教程可能过期了,请直接看最新版(perl模块安装大全)
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
前面我简单写了一个perl的cpan安装模块,但是前些天突然发现有些perl模块在cpan里面找不到,所以又总结了一下不同的perl模块安装方法。 Continue reading
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
在我的第一讲里面,对JAVA的安装,其实就需要添加环境变量,大家可以回头看看!
添加PATH环境变量,第1种方法:
[root@lx_web_s1 ~]# export PATH=/usr/local/webserver/mysql/bin:$PATH
再次查看:
[root@lx_web_s1 ~]# echo $PATH
/usr/local/webserver/mysql/bin:/usr/local/webserver/mysql/bin/:/usr/kerberos/sbin:/usr/kerberos/bin:/usr/local/sbin:/usr/local/bin:/sbin:/bin:/usr/sbin:/usr/bin:/root/bin
说明添加PATH成功。
上述方法的PATH 在终端关闭 后就会消失。所以还是建议通过编辑/etc/profile来改PATH,也可以修改家目录下的.bashrc(即:~/.bashrc)。
第2种方法:需要管理员权限。
# vim /etc/profile
在最后,添加:
export PATH="/usr/local/webserver/mysql/bin:$PATH"
保存,退出,然后运行:
#source /etc/profile,不报错则成功。
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
同样,这个ssh也非常简单
sudo apt-get install openssh-server
SSH分客户端openssh-client和openssh-server
如果你只是想登陆别的机器的SSH只需要安装openssh-client(ubuntu有默认安装,如果没有则sudo
apt-get install openssh-client),如果要使本机开放SSH服务就需要安装openssh-server
sudo apt-get install openssh-server
然后确认sshserver是否启动了:
ps -e |grep ssh
如果看到sshd那说明ssh-server已经启动了。
如果没有则可以这样启动:sudo /etc/init.d/ssh start 或者 service ssh start
ssh-server配置文件位于/etc/ssh/sshd_config,在这里可以定义SSH的服务端口,默认端口是22,你可以自己定义成其他端口号,如222。
然后重启SSH服务:
sudo
/etc/init.d/ssh stop
sudo /etc/init.d/ssh start
然后使用以下方式登陆SSH:
ssh username@192.168.1.112 username为192.168.1.112 机器上的用户,需要输入密码。
我给七八个虚拟机都配置成功了,但是呢,偏偏别人的一个我始终不能解决,感觉linux里面的学问还是蛮多的
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
这个主要是针对有界面的服务器来说的,不是我们通常意义的ssh登陆,一般ssh登陆的可以把中文复制张贴进去即可。
Ubuntu上的输入法主要有小小输入平台(支持拼音/二笔/五笔等),Fcitx,Ibus,Scim等。其中Scim和Ibus是输入法框架。
在Ubuntu的中文系统中自带了中文输入法,通过Ctrl+Space可切换中英文输入法。这里我们主要说下Ubuntu英文系统中,中文输入法的安装。
安装输入法的第一步,是安装语言包。我们选择System Settings-->Language Support-->Install/Remove Languages,这里面可以选择简体中文
输入密码后,系统会安装简体中文语言包。
第二步,安装完毕后切换到终端,安装IBus框架,在终端输入以下命令:
sudo apt-get install ibus ibus-clutter ibus-gtk ibus-gtk3 ibus-qt4
启动IBus框架,在终端输入:
im-switch -s ibus
安装完IBus框架后注销系统,保证更改立即生效。
第三步:安装拼音引擎
有下面几种常用选择:
IBus拼音:sudo apt-get install ibus-pinyin
IBUS五笔:sudo apt-get install ibus-table-wubi
谷歌拼音输入法:sudo apt-get install ibus-googlepinyin
Sun拼音输入法:sudo apt-get install ibus-sunpinyin
第四步:设置IBus框架
终端输入ibus-setup 此时,IBus Preference设置被打开。我们在Input Method选项卡中,选择自己喜欢的输入方式,并配置自己喜欢的快捷键即可。
第五步:通常情况下,IBus图标(一个小键盘)会出现在桌面右上角的任务栏中。有时候这个图标会自行消失,可使用以下命令,找回消失的IBus图标:
ibus-daemon –drx
发现自己搞服务器遇到的困难还是蛮多的,所以记录了一下,给菜鸟们指个路。
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
理论上perl是不需要更新,但是我就不巧碰到了这个情况,所以也记录一下
linux下升级系统默认安装的perl版本,不建议先rm
先下载tar.gz ...然後手动安装..default 安装到/usr/local/目录下..
然後修改/usr/bin/perl的symbolic link到/usr/local/bin/perl
下载方式不用说了吧,各显神通,笔者习惯用wget.
所以wget[url]http://www.cpan.org/src/perl-5.10.0.tar.gz[/url] .现在最新是5.20
下载完以后解压安装
#tar zxvf perl-5.10.0.tar.gz
#cd perl-5.10.0
#./Configure -des -Dprefix=/usr/local/perl
参数-Dprefix指定安装目录为/usr/local/perl
#make
#make test
#make install
如果这个过程没有错误的话,那么恭喜你安装完成了.是不是很简单?
接下来替换系统原有的perl,有最新的了咱就用嘛.
#mv /usr/bin/perl/ usr/bin/perl.bak
#ln -s /usr/local/perl/bin/perl/ usr/bin/perl
#perl –v
然后就可以了用它来安装一些其它你需要的perl模块了
#perl -MCPAN-e shell
第一次执行的话,会提示安装cpan并要求连接网络下载最新的模块列表.然后就可以安装东西了
cpan[1]> install DBI
)
ubuntu对生信菜鸟来说是最好用的linux服务器,没有之一,因为它有apt-get。
1、JDK官网上http://www.oracle.com/technetwork/java/javase/downloads/index.html选择:
但是,如果你的服务器是64位的,请不要选择i586,选择你自己的机器对应的!
2、将打开终端,建立目录:
Sudo mkdir /usr/lib/java
3、将下载的 jdk-7u3-linux-i586.tar.gz移到这个文件夹下面并进行解压,改名字:
sudo mv jdk-7u3-linux-i586.tar.gz /usr/lib/java
sudo tar –xvf jdk-7u3-linux-i586.tar.gz
mv jdk1.7.0_03java-7-sun
4、修改环境变量:
在终端输入:vim /etc/profile
然后添加以下代码:
export JAVA_HOME=/usr/lib/java/jdk1.8.0_25
export JRE_HOME=${JAVA_HOME}/jre
export CLASSPATH=.:${JAVA_HOME}/lib:${JRE_HOME}/lib
export PATH=${JAVA_HOME}/bin:$PATH
保存之后,再运行下面命令更新电脑的配置文件
source /etc/profile 这个千万要记得!!!!
5、在终端中输入 java –version,显示:
jeydragon@jeydragon-VirtualBox:~$ java -version
java version "1.7.0_03"
Java(TM) SE Runtime Environment (build 1.7.0_03-b04)
Java HotSpot(TM) Client VM (build 22.1-b02, mixed mode)
表示安装成功
之所以要讲一下这个,是因为trinity这个软件居然需要perl的模块才能使用,所以我必须自己安装几个模块。此教程可能过期了,请直接看最新版(perl模块安装大全)
前提是要有root权限,否则只能自己下载perl模块自己解压安装了。 Continue reading
刚买了一个空的云服务器,所以就试用体验了一下!
一.硬盘容量,系统盘很小,就30个G,但是有一个1T的空盘,自己格式化安装并挂载即可。
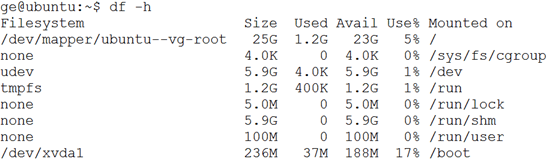
二.内存状况,11G

三。Cup数量,12核

四.磁盘文件状况

五.开通其它账号
adduser
六.服务器其它信息
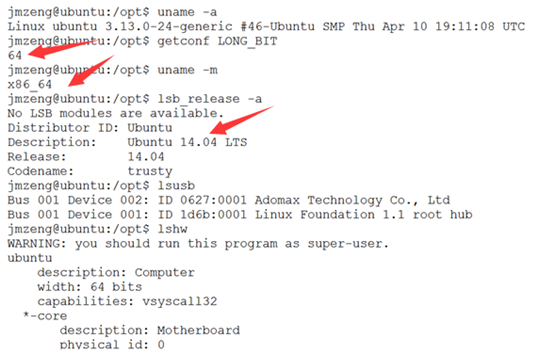
七.测试下载速度
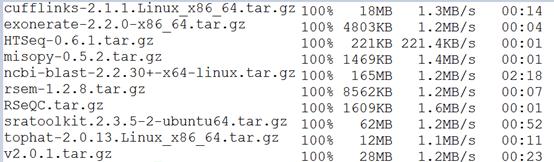
八.磁盘分区管理
首先看到的是32.2GB的系统盘,是标示符是xvda盘,被分成了三个区
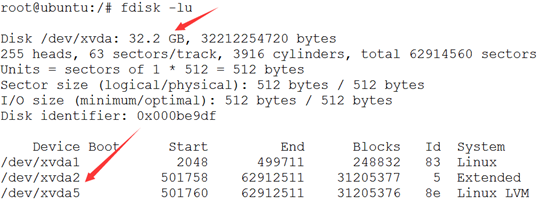
然后是一个1T的硬盘,需要分区然后挂载
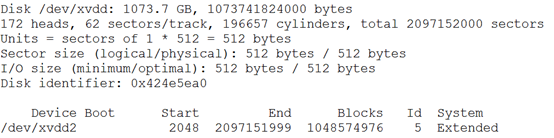
我分区后,分割成两个盘,一个给各个用户,还有一个放公共数据
apt-get install nfs-common
mkfs -t ext4 /dev/xvdd
mkfs.ext4 /dev/xvdd1
mount -t ext4 /dev/xvdd1 /home/
mount -t ext4 /dev/xvdd2 /data
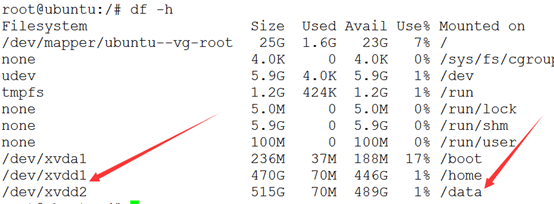
九.脚本环境
This is perl 5, version 18, subversion 2 (v5.18.2)
/usr/lib/python2.7/site.pyc matches /usr/lib/python2.7/site.py
十.库文件状况
apt-get install unzip
apt-get install make
apt-get install gcc
十一.软件安装状况
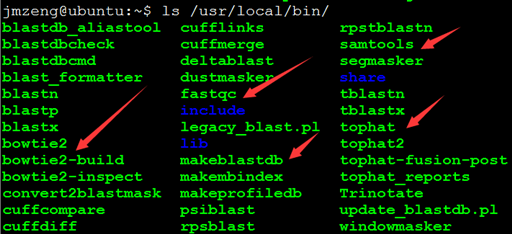
常用生物信息学软件都可以自己安装,并且可以使用。
查询需要根据前面建立的索引来做。
这是一个比较复杂的过程,我也是看了bowtie的作者的ppt才慢慢弄懂的,感觉自己也不可能三言两语就说清楚,一般都是辅助图片,动画,再经过多方交流才能慢慢理解。
所以大家呢,就自己去看ppt,看懂那个查询算法。(ppt及代码在我的群里面有共享,欢迎大家加群交流)
这里我简单讲讲我的程序
首先读取索引文件,统计好A,C,G,T的总数
然后把查询序列从最后一个字符往前面回溯。
我创建了一个子函数,专门来处理回溯的问题
每次接受四个参数(左右两端的碱基,上下的阈值),并返回两个参数(新的上下两个阈值)
大家要看懂阈值是如何更新迭代,这样动态的一个个回溯字符串,一个个迭代阈值。
直到四种临界情况的出现。
第一是上下阈值已经相等了,但是我们还没有回溯完全,那就说明字符串只能查找后几个字符,前面还有字符是无法匹配的
第二种情况是上下阈值已经相等了,正巧我们也回溯到了最后一个字符串,那么我们就找到了精确匹配。
第三种情况是已经进行到了最后一个字符串,但是上下阈值还有差值,那么就找到了多个精确匹配点。
最后一种情况是各种非法字符。
然后我简单的测序了一下在病毒的5K基因组里面的精确匹配情况,好像效果还挺好的
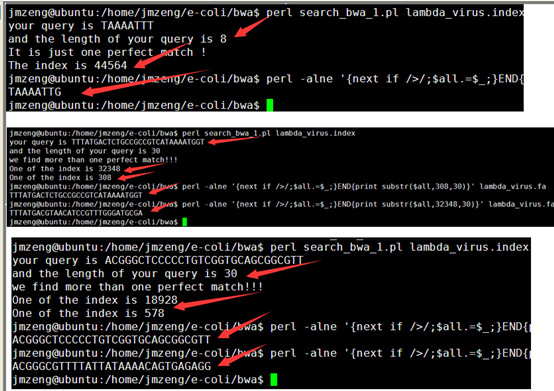
但是在酵母里面还有一个问题没有解决,就是取前二十个字符串排序的问题,不够精确,需要重新审视排序结果进行局部优化,可能是需要用堆排序发,具体我还得考虑一个星期,只能等下周上课再看看了,平时太忙了,基本没时间码代码。
这里贴上我的代码给大家看看,
[perl]
$a='CGCTATGTACTGGATGCGCTGGCAAACGAGCCTGCCGTAAG';
while(<>){
chomp;
@F=split;
$hash_count_atcg{$F[0]}++;
$hash{$.}=$_;
}
$all_a=$hash_count_atcg{'A'};
$all_c=$hash_count_atcg{'C'};
$all_g=$hash_count_atcg{'G'};
$all_t=$hash_count_atcg{'T'};
#print "$all_a\t$all_c\t$all_g\t$all_t\n";
$len_a=length $a;
$end_a=$len_a-1;
print "your query is $a\n";
print "and the length of your query is $len_a \n";
foreach (reverse (0..$end_a)){
$after=substr($a,$_,1);
$before=substr($a,$_-1,1);
#对第一个字符进行找阈值的时候,我们需要人为的定义起始点!
if($_ == $end_a){
if ($after eq 'A') {
$start=1;
$end=$all_a;
}
elsif ($after eq 'C') {
$start=$all_a+1;
$end=$all_a+$all_c;
}
elsif ($after eq 'G') {
$start=$all_a+$all_c+1;
$end=$all_a+$all_c+$all_g;
}
elsif ($after eq 'T'){
$start=$all_a+$all_c+$all_g+1;
$end=$all_a+$all_c+$all_g+$all_t;
}
else {print "error !!! we just need A T C G !!!\n";exit;}
}
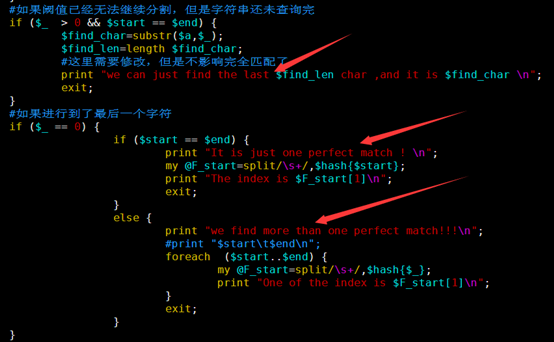
#如果阈值已经无法继续分割,但是字符串还未查询完
if ($_ > 0 && $start == $end) {
$find_char=substr($a,$_);
$find_len=length $find_char;
#这里需要修改,但是不影响完全匹配了
print "we can just find the last $find_len char ,and it is $find_char \n";
exit;
}
#如果进行到了最后一个字符
if ($_ == 0) {
if ($start == $end) {
print "It is just one perfect match ! \n";
my @F_start=split/\s+/,$hash{$start};
print "The index is $F_start[1]\n";
exit;
}
else {
print "we find more than one perfect match!!!\n";
#print "$start\t$end\n";
foreach ($start..$end) {
my @F_start=split/\s+/,$hash{$_};
print "One of the index is $F_start[1]\n";
}
exit;
}
}
($start,$end)=&find_level($after,$before,$start,$end);
}
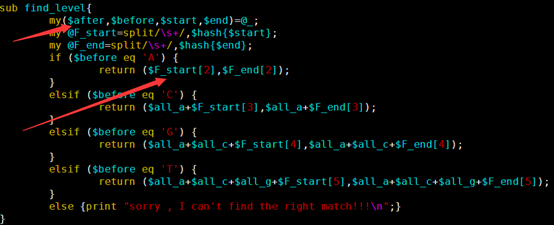
sub find_level{
my($after,$before,$start,$end)=@_;
my @F_start=split/\s+/,$hash{$start};
my @F_end=split/\s+/,$hash{$end};
if ($before eq 'A') {
return ($F_start[2],$F_end[2]);
}
elsif ($before eq 'C') {
return ($all_a+$F_start[3],$all_a+$F_end[3]);
}
elsif ($before eq 'G') {
return ($all_a+$all_c+$F_start[4],$all_a+$all_c+$F_end[4]);
}
elsif ($before eq 'T') {
return ($all_a+$all_c+$all_g+$F_start[5],$all_a+$all_c+$all_g+$F_end[5]);
}
else {print "sorry , I can't find the right match!!!\n";}
}
#perl -alne '{next if />/;$all.=$_;}END{print substr($all,308,10)}' lambda_virus.fa
[/perl]
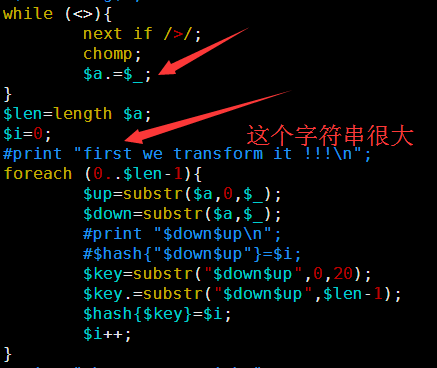
其中第一讲我提到了一个简单的索引产生方式,因为是课堂就半个小时想的,很多细节没有考虑到,对病毒那种几K大小的基因组来说是很简单的,速度也非常快,但是我测试了一下酵母,却发现好几个小时都没有结果,我只好kill掉重新改写算法,我发现之前的测序最大的问题在于没有立即substr函数的实现方式,把一个5M的字符串不停的截取首尾字符串好像是一个非常慢的方式。
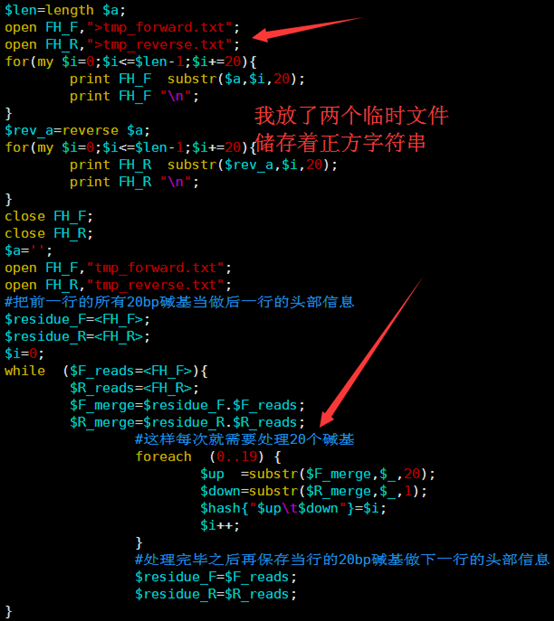
所以我优化了那个字符串的函数,虽然代码量变多了,实现过程也繁琐了一点,但是速度提升了几千倍。
time perl bwt_new_index.pl e-coli.fa >e-coli.index
测试了一下我的脚本,对酵母这样的5M的基因组,索引耗费时间是43秒
real 0m43.071s
user 0m41.277s
sys 0m1.779s
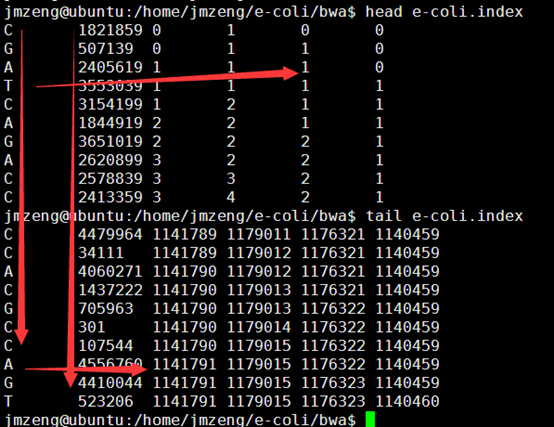
输出的index矩阵如下,我简单的截取头尾各10行给大家看,一点要看懂这个index。
首先第一列就是我们的BWT向量,也就是BWT变换后的尾字符
第二列是之前的顺序被BWT变换后的首字符排序后的打乱的顺序。
第三,四,五,六列分别是A,C,G,T的计数,就是在当行之前累积出现的A,C,G,T的数量,是对第一列的统计。
这个索引文件将会用于下一步的查询,这里贴上我新的索引代码,查询见下一篇文章
[perl]
while (<>){
next if />/;
chomp;
$a.=$_;
}
$len=length $a;
open FH_F,">tmp_forward.txt";
open FH_R,">tmp_reverse.txt";
for(my $i=0;$i<=$len-1;$i+=20){
print FH_F substr($a,$i,20);
print FH_F "\n";
}
$rev_a=reverse $a;
for(my $i=0;$i<=$len-1;$i+=20){
print FH_R substr($rev_a,$i,20);
print FH_R "\n";
}
close FH_F;
close FH_R;
$a='';
open FH_F,"tmp_forward.txt";
open FH_R,"tmp_reverse.txt";
#把前一行的所有20bp碱基当做后一行的头部信息
$residue_F=<FH_F>;
$residue_R=<FH_R>;
$i=0;
while ($F_reads=<FH_F>){
$R_reads=<FH_R>;
$F_merge=$residue_F.$F_reads;
$R_merge=$residue_R.$R_reads;
#这样每次就需要处理20个碱基
foreach (0..19) {
$up =substr($F_merge,$_,20);
$down=substr($R_merge,$_,1);
$hash{"$up\t$down"}=$i;
$i++;
}
#处理完毕之后再保存当行的20bp碱基做下一行的头部信息
$residue_F=$F_reads;
$residue_R=$R_reads;
}
#print "then we sort it\n";
$count_a=0;
$count_c=0;
$count_g=0;
$count_t=0;
foreach (sort keys %hash){
$first=substr($_,0,1);
$len=length;
$last=substr($_,$len-1,1);
#print "$first\t$last\t$hash{$_}\n";
$count_a++ if $last eq 'A';
$count_c++ if $last eq 'C';
$count_g++ if $last eq 'G';
$count_t++ if $last eq 'T';
print "$last\t$hash{$_}\t$count_a\t$count_c\t$count_g\t$count_t\n";
}
unlink("tmp_forward.txt");
unlink("tmp_reverse.txt");
[/perl]
一、下载及安装软件
这个软件需要edu邮箱注册才能下载,可能是仅对科研高校开放吧。所以软件地址我就不列了。
它其实是几个perl程序,比较重要的是这个人类的数据库,snp注释必须的。
参考:http://annovar.readthedocs.org/en/latest/misc/accessory/
二,准备数据
既然是注释,那当然要有数据库啦!数据库倒是有下载地址
http://www.openbioinformatics.org/annovar/download/hg19_ALL.sites.2010_11.txt.gz
也可以用命令来下载
Perl ./annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
然后我们是对snp-calling流程跑出来的VCF文件进行注释,所以必须要有自己的VCF文件,VCF格式详解见本博客另一篇文章,或者搜索也行
http://vcftools.sourceforge.net/man_latest.html
三、运行的命令
首先把vcf格式文件,转换成空格分隔格式文件,自己写脚本也很好弄
perl convert2annovar.pl -format vcf
/home/jmzeng/raw-reads/whole-exon/snp-calling/tmp1.vcf >annovar.input
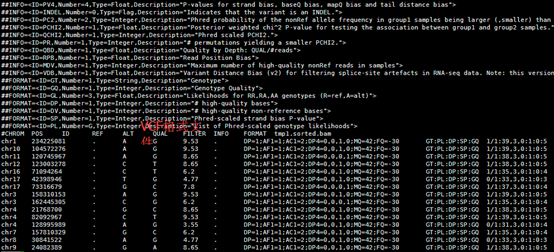
变成了空格分隔的文件
然后把转换好的数据进行注释即可
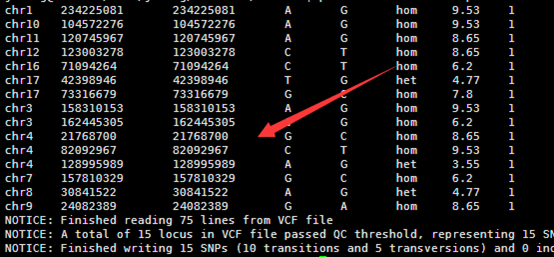
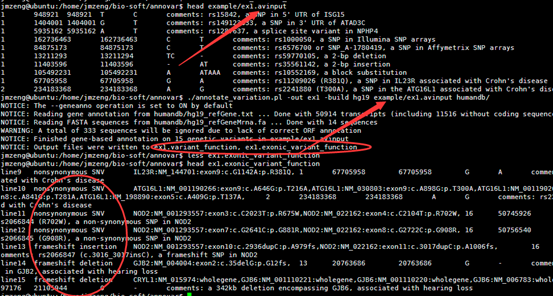
./annotate_variation.pl -out ex1 -build hg19 example/ex1.avinput humandb/
四,输出文件解读
比对可以选择BWA或者bowtie,测序数据可以是单端也可以是双端,我这里简单讲一个,但是脚本都列出来了。而且我选择的是bowtie比对,然后单端数据。
首先进入hg19的目录,对它进行两个索引
samtools faidx hg19.fa
Bowtie2-build hg19.fa hg19
我这里随便从26G的测序数据里面选取了前1000行做了一个tmp.fa文件,进入tmp.fa这个文件的目录进行操作
Bowtie的使用方法详解见http://www.bio-info-trainee.com/?p=398
bowtie2 -x ../../../ref-database/hg19 -U tmp1.fa -S tmp1.sam
samtools view -bS tmp1.sam > tmp1.bam
samtools sort tmp1.bam tmp1.sorted
samtools index tmp1.sorted.bam
samtools mpileup -d 1000 -gSDf ../../../ref-database/hg19.fa tmp1.sorted.bam |bcftools view -cvNg - >tmp1.vcf
然后就能看到我们产生的vcf变异格式文件啦!
当然,我们可能还需要对VCF文件进行再注释!
要看懂以上流程及命令,需要掌握BWA,bowtie,samtools,bcftools,
数据格式fasta,fastq,sam,vcf,pileup
如果是bwa把参考基因组索引化,然后aln得到后缀树,然后sampe对双端数据进行比对
首先bwa index 然后选择算法,进行索引。
然后aln脚本批量处理
==> bwa_aln.sh <==
while read id
do
echo $id
bwa aln hg19.fa $id >$id.sai
done <$1
然后sampe脚本批量处理
==> bwa_sampe.sh <==
while read id
do
echo $id
bwa sampe hg19.fa $id*sai $id*single >$id.sam
done <$1
然后是samtools的脚本
==> samtools.sh <==
while read id
do
echo $id
samtools view -bS $id.sam > $id.bam
samtools sort $id.bam $id.sorted
samtools index $id.sorted.bam
done <$1
然后是bcftools的脚本
==> bcftools.sh <==
while read id
do
echo $id
samtools mpileup -d 1000 -gSDf ref.fa $id*sorted.bam |bcftools view -cvNg - >$id.vcf
done <$1
==> mpileup.sh <==
while read id
do
echo $id
samtools mpileup -d 100000 -f hg19.fa $id*sorted.bam >$id.mpileup
done <$1
需要插件和自己修改主题下面的foot.php代码。
参考 http://jingyan.baidu.com/article/ae97a646ce37c2bbfd461d01.html
步骤如下:
1、登陆到wp后台,鼠标移动到左侧菜单的“插件”链接上,会弹出子菜单,点击子菜单的“安装插件”链接
2、WP-PostViews插件显示wordpress文章点击浏览量
在“安装插件”链接页面的搜索框中输入“WP-PostViews”,然后回车
3、WP-PostViews插件显示wordpress文章点击浏览量
在搜索结果页面点击“WP-PostViews”插件内容区域的“现在安装”按钮
4、WP-PostViews插件显示wordpress文章点击浏览量
程序自动下载插件到服务器并解压安装,一直等到安装成功信息出现,然后在安装成功提示页面点击“启动插件”链接。
5、WP-PostViews插件显示wordpress文章点击浏览量
页面会自动跳转到“已安装插件”页面,在已安装插件列表中我们可以看到“Form Manager”插件已经处于启用状态(插件名下是“停用”链接)。
有了这个插件之后,我们的整个网页环境里面就多了一个 the_views()函数,它统计着每个文章的点击数,这样我们之前的网页就能显示点击数了。
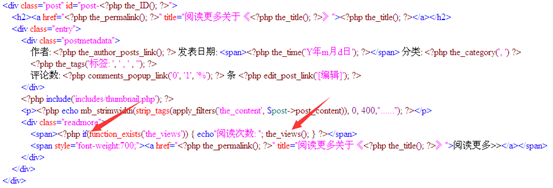
这个是我现在用的主题的php代码,把文章用span标记隔开了,而且显示着上面php代码里面的每一个内容包括日期,分类,标签,评论等等
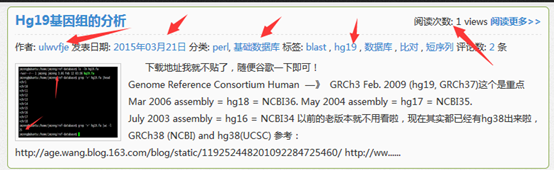
其中thez-view()这个函数返回的不仅仅是一个访客数,但是我的文章的访客都太少了,所以我写了一个脚本帮我刷一刷流量。
[perl]
use List::MoreUtils qw(uniq);
$page='http://www.bio-info-trainee.com/?paged=';
foreach (1..5){ #我的文章比较少,就42个,所以只有5个页面
$url_page=$page.$_;
$tmp=`curl $url_page`;
#@p=$tmp=~/p=(\d+)/;
$tmp =~ s/(p=\d+)/push @p, $1/eg; #寻找p=数字这样的标签组合成新的网页地址
}
@p=uniq @p;
print "$_\n" foreach @p; #可以找到所有42个网页的地址
foreach (@p){
$new_url='http://www.bio-info-trainee.com/?'.$_;
`curl $new_url` foreach (1..100); #每个网页刷一百次
}
[/perl]
大家可以看到这个网页被刷的过程,从15到21到27直到100
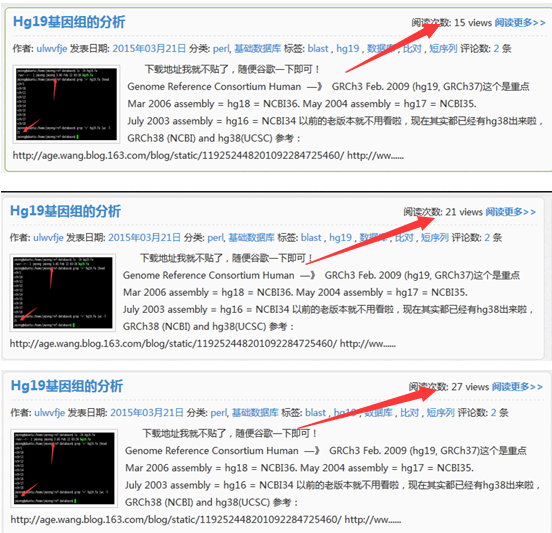
大家现在再去看我的网页,就每个文章都有一百的访问量啦!
http://www.bio-info-trainee.com/
下载地址我就不贴了,随便谷歌一下即可!
Genome Reference Consortium Human ---》 GRCh3
Feb. 2009 (hg19, GRCh37)这个是重点
Mar 2006 assembly = hg18 = NCBI36.
May 2004 assembly = hg17 = NCBI35.
July 2003 assembly = hg16 = NCBI34
以前的老版本就不用看啦,现在其实都已经有hg38出来啦,GRCh38 (NCBI) and hg38(UCSC)
参考:http://age.wang.blog.163.com/blog/static/119252448201092284725460/
http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/
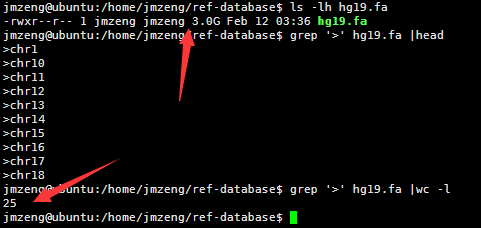
人的hg19基因组是3G的大小,因为一个英文字符是一个字节,所以也是30亿bp的碱基。
包括22条常染色体和X,Y性染色体及M线粒体染色体。
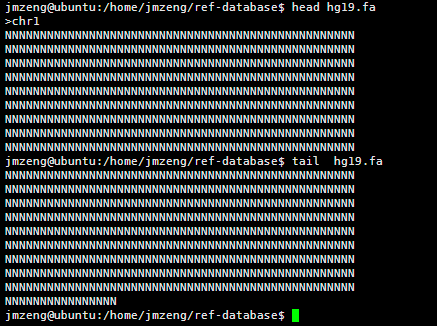
查看该文件可以看到,里面有很多的N,这是基因组里面未知的序列,用N占位,但是觉得部分都是A.T.C.G这样的字符,大小写都有,分别代表不同的意思。
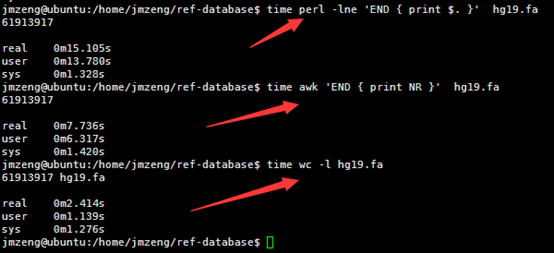
然后我用linux的命令统计了一下里面这个文件的行数,
perl -lne 'END { print $. }' hg19.fa
awk 'END { print NR }' hg19.fa
wc -l hg19.fa
然后我写了一个脚本统计每条染色体的长度,42秒钟完成任务!
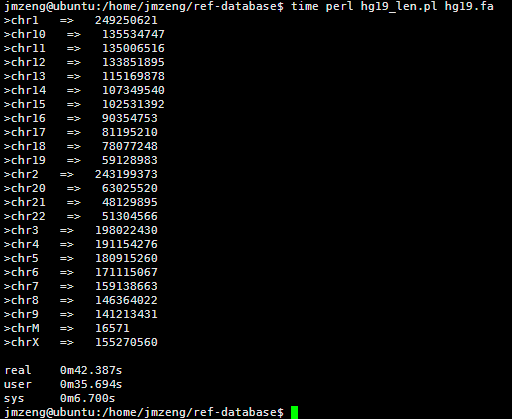
看来这个服务器的性能还是蛮强大的,读取文件非常快!
[perl]
while(<>){
chomp;
if (/>/){
if (exists $hash_chr{$key} ){
$len = length $hash_chr{$key};
print "$key => $len\n";
}
undef %hash_chr;
$key=$_;
}
else {
$hash_chr{$key}.=$_;
}
}
[/perl]
然后我用seed统计了一下hg19的词频(我不知道生物信息学里面的专业描述词语是什么)
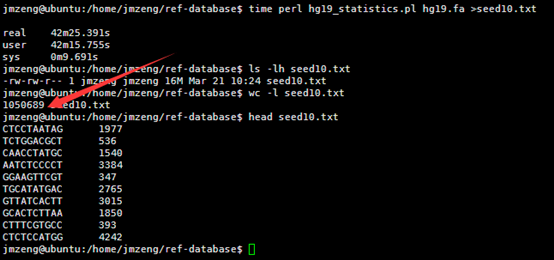
我的程序耗费了42分钟才跑完,感觉我写的程序应该是没有问题的,让我吃惊的是总共竟然只有105万条独特的10bp短序列。然后我算了一下4的10次方,(⊙o⊙)…悲剧,原来只有1048576,之所以出现这种情况,是因为里面有N这个字符串,不仅仅是A.T.C.G四个字符。我用grep -v N seed10.txt |wc -l命令再次统计了一下,发现居然就是1048576,也就是说,任意A.T.C.G四个字符组成的10bp字符串短序列在人的基因组里面都可以找到!!!
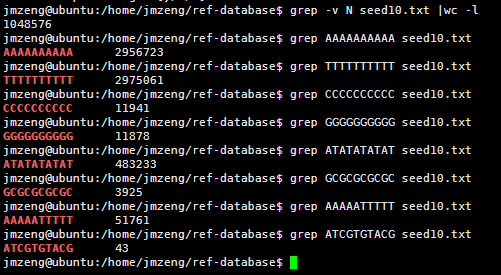
然后我测试了一下,还是真是这样的,真是一个蛮有意思的现象。虽然我无法解释为什么,但是根据这个结果我们可以得知连续的A或者T在人类基因组里面高频出现,而连续的G或者C却很少!
如果我们储存这个10bp字符串的同时,也储存着它们在基因组的位置,那么就可以根据这个seed来进行比对,这就是blast的原理之一!
以前的一下perl代码分享
今天去参加了开源中国的一个源创会,感觉好隆重的样子,近五百人,BAT的工程师都过来演讲了,可都是数据库相关的, 我一个的都没有听懂,但是茶歇的披萨我倒是吃了不少。
说到开源中国,我想起来了我以前在上面分享的代码,上去看了看,竟然有那么多的访问量了,让我蛮意外的,那些代码完全是我学习perl的历程的真实写照。
想了想,既然是菜鸟教程,那就索性再介绍点更基础的东西,基本上只要是大学毕业的都能看懂,不需要懂计算机了。首先讲讲linux服务器吧,因为生物信息也算是半个大数据分析,所以我们平常的办公电脑一般都是不能满足需求的,大部分实验室及公司都会自己配置好服务器给菜鸟们用,菜鸟们首先要拿到服务器的IP和高手给你的用户名和密码。
一般我们讲服务器,大多是linux系统,而我这里所讲的linux系统呢,特指ubuntu,其余的我懒得管了,大家也不要耗费无谓的时间纠结那些名词的不同!
登录到服务器有两种方法,一种是ssh,传输你的命令给服务器执行,另一种是ftp,和服务器交换文件。而ssh我们通常用putty,xshell等等。ftp呢,我们可以用winscp,xshell,所以我一直都用xshell,因为它两者都能搞定!
Xshell软件自行搜索下载,打开之后新建一个连接,然后登陆即可。
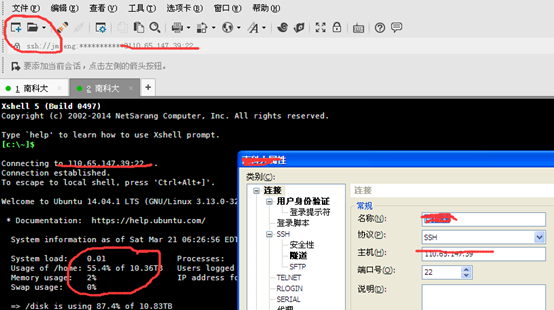
然后输入以下命令,可以查看服务器配置,包括cpu。内存,还有硬盘
cat /proc/cpuinfo |grep pro|wc -l
free -g
df -h
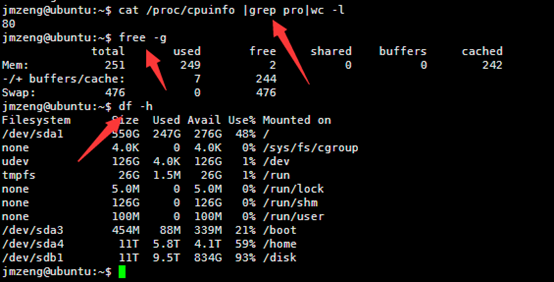
这个服务器配置好一点,有80个cpu,内存256G,硬盘有2个11T的,是比较成熟的配置。
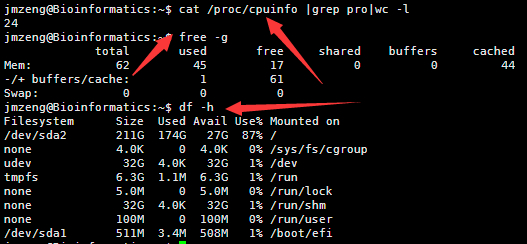
这个是一个小型服务器。也就24个核,64G的内存,但是存储量有点小呀,其实可以随便花几百块钱买个1T的硬盘挂载上去的。
然后linux的其它命令大家就得自己去搜索一个个使用,然后熟悉,记牢,然后创新啦!
我随便敲几个我常用的吧: ls cd mkdir rm cp cat head tail more less diff grep awk sed grep perl 等等!
呀,突然间发现我才介绍了ssh的方法登陆服务器并且发送命令在服务器上面运行,下面贴图如何传输文件。一般xshell的菜单里面有绿的文件夹形式的标签就是打开ftp文件传输,这种可视化的软件,大家慢慢摸索吧!