这个技巧很重要,一般来说,R语言自带的install.packages函数来安装一个包时,都是用的默认的镜像!
如果你是用的Rstudio这个IDE,你的默认镜像就是: https://cran.rstudio.com/
如果你直接用的R语言,那么就是:"http://cran.us.r-project.org" 但是一般你安装的时候会提醒你选择。
这个技巧很重要,一般来说,R语言自带的install.packages函数来安装一个包时,都是用的默认的镜像!
如果你是用的Rstudio这个IDE,你的默认镜像就是: https://cran.rstudio.com/
如果你直接用的R语言,那么就是:"http://cran.us.r-project.org" 但是一般你安装的时候会提醒你选择。
既然你点进来看,肯定是有需求咯! 一般来说,R语言自带的install.packages函数来安装一个包时,都是默认安装最新版的。 但是有些R包的开发者他会引用其它的一些R包,但是它用的是人家旧版本的功能,但他自己来不及更新或者疏忽了。 而我们又不得不用他的包,这时候就不得不卸载最新版包,转而安装旧版本包。
limma真不愧是最流行的差异分析包,十多年过去了,一直是芯片数据处理的好帮手。
现在又可以支持RNA-seq数据,我赶紧试用了一下!
我下面只讲用法,大家看代码就明白了!
最近经常出现一个错误,类似于package ‘airway’ is not available (for R version 3.1.0)
就是某些包在R的仓库里面找不到,这个错误非常普遍,stackoverflow上面非常详细的解答:
在阅读这个答案的时候,我发现了一个非常有用的函数!available.packages()可以查看自己的机器可以安装哪些包!
我以前写过DESeq,以及过时了:http://www.bio-info-trainee.com/867.html
正好准备筹集bioconductor中文社区,我写简单讲一下DESeq2这个包如何用!
shiny是Rstudio公司推出的一个网页服务器,可以直接用R语言制作客户端和服务端程序来交互式的展现数据,而且有越来越丰富的扩展包可以借鉴使用,我觉得它的前途很光明,值得大家入坑!
虽然Rstudio公司吹嘘初学者无需具备html和css,js基础,但是个人认为还是多了解一下比较好,可以在w3cschool里面在线学习!
新手安装好shiny包之后里面自带了12个app例子,一边调试一边学习,很快就可以入门了。推荐一个教程,用shiny构建网页中文教程,想查看更详细的介绍和实例,请访问Shiny的官方主页。当然,你肯定需要要会R,这里有个R的FAQ教程。
如果你完全看完了上面那些,说明你已经真正入门了!
你现在可以去搜索一个shiny cheetsheet,然后背诵下来!!!!
然后你可以去shiny的github里面找到108个示例程序,一个个运行了解它们!!!
这时候你已经是高手啦!!!
接下来你应该取了解一个叫做dashboard的东西,熟练UI界面设计。
最后你再了解一些辅助包,帮助你用shiny更好的展示数据,主要是JS绘图插件
里面最出名的就是DT包了,一定要学!!!
最后你可以了解一下在rstudio上面分享五个免费的shiny网页程序,需要你搜索一些。
如果你还感兴趣的话,就可以自己整一个虚拟机Ubuntu或者centos系统,试着安装shiny的server,这个比较考验技术。
总结:你可以需要200个小时左右来完全掌握shiny
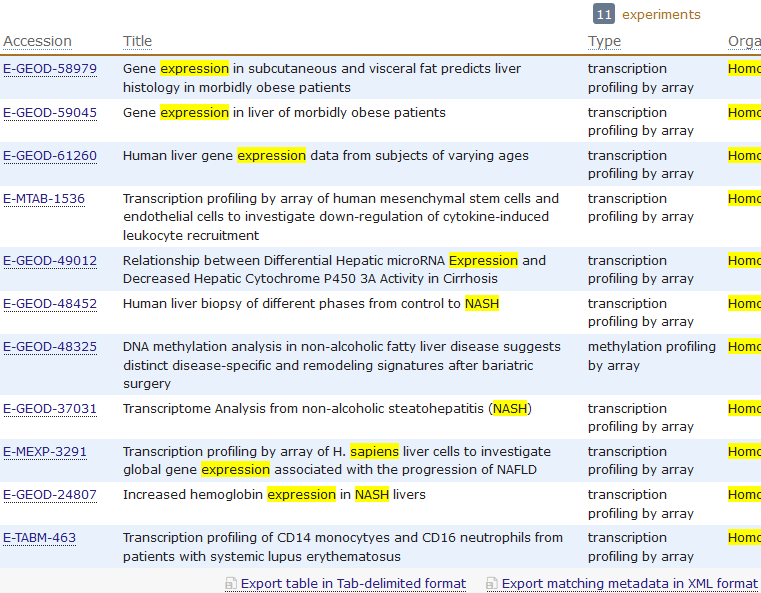
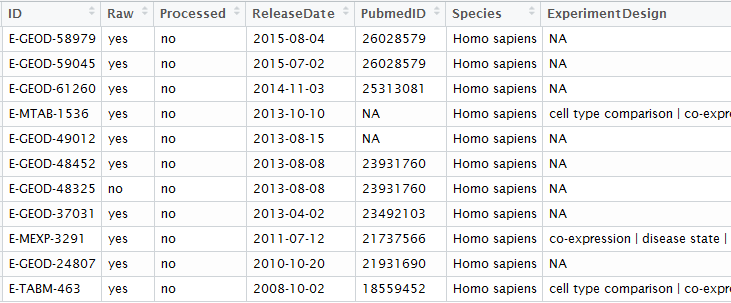
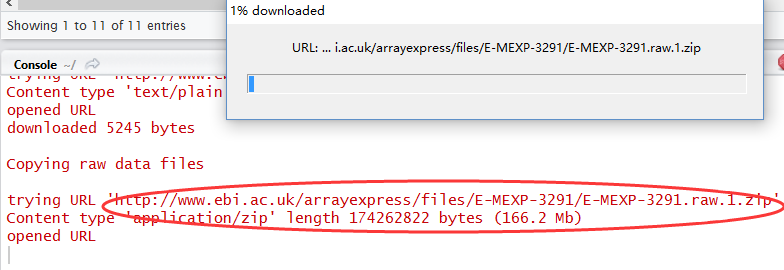
rawset = ArrayExpress("E-MEXP-3291")
现有的基因芯片种类不要太多了!
gpl organism bioc_package1 GPL32 Mus musculus mgu74a2 GPL33 Mus musculus mgu74b3 GPL34 Mus musculus mgu74c6 GPL74 Homo sapiens hcg1107 GPL75 Mus musculus mu11ksuba8 GPL76 Mus musculus mu11ksubb9 GPL77 Mus musculus mu19ksuba10 GPL78 Mus musculus mu19ksubb11 GPL79 Mus musculus mu19ksubc12 GPL80 Homo sapiens hu680013 GPL81 Mus musculus mgu74av214 GPL82 Mus musculus mgu74bv215 GPL83 Mus musculus mgu74cv216 GPL85 Rattus norvegicus rgu34a17 GPL86 Rattus norvegicus rgu34b18 GPL87 Rattus norvegicus rgu34c19 GPL88 Rattus norvegicus rnu3420 GPL89 Rattus norvegicus rtu3422 GPL91 Homo sapiens hgu95av223 GPL92 Homo sapiens hgu95b24 GPL93 Homo sapiens hgu95c25 GPL94 Homo sapiens hgu95d26 GPL95 Homo sapiens hgu95e27 GPL96 Homo sapiens hgu133a28 GPL97 Homo sapiens hgu133b29 GPL98 Homo sapiens hu35ksuba30 GPL99 Homo sapiens hu35ksubb31 GPL100 Homo sapiens hu35ksubc32 GPL101 Homo sapiens hu35ksubd36 GPL201 Homo sapiens hgfocus37 GPL339 Mus musculus moe430a38 GPL340 Mus musculus mouse430239 GPL341 Rattus norvegicus rae230a40 GPL342 Rattus norvegicus rae230b41 GPL570 Homo sapiens hgu133plus242 GPL571 Homo sapiens hgu133a243 GPL886 Homo sapiens hgug4111a44 GPL887 Homo sapiens hgug4110b45 GPL1261 Mus musculus mouse430a249 GPL1352 Homo sapiens u133x3p50 GPL1355 Rattus norvegicus rat230251 GPL1708 Homo sapiens hgug4112a54 GPL2891 Homo sapiens h20kcod55 GPL2898 Rattus norvegicus adme16cod60 GPL3921 Homo sapiens hthgu133a63 GPL4191 Homo sapiens h10kcod64 GPL5689 Homo sapiens hgug4100a65 GPL6097 Homo sapiens illuminaHumanv166 GPL6102 Homo sapiens illuminaHumanv267 GPL6244 Homo sapiens hugene10sttranscriptcluster68 GPL6947 Homo sapiens illuminaHumanv369 GPL8300 Homo sapiens hgu95av270 GPL8490 Homo sapiens IlluminaHumanMethylation27k71 GPL10558 Homo sapiens illuminaHumanv472 GPL11532 Homo sapiens hugene11sttranscriptcluster73 GPL13497 Homo sapiens HsAgilentDesign02665274 GPL13534 Homo sapiens IlluminaHumanMethylation450k75 GPL13667 Homo sapiens hgu21976 GPL15380 Homo sapiens GGHumanMethCancerPanelv177 GPL15396 Homo sapiens hthgu133b78 GPL17897 Homo sapiens hthgu133a
gpl_info=read.csv("GPL_info.csv",stringsAsFactors = F)### first download all of the annotation packages from bioconductorfor (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform) ##主要是因为我处理包的字符串前面有空格#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) == FALSE) {BiocInstaller::biocLite(platformDB)#source("http://bioconductor.org/biocLite.R");#biocLite(platformDB )}}
下载完了所有的包, 就可以进行批量导出芯片探针与gene的对应关系!
for (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform)#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) != FALSE) {library(platformDB,character.only = T)#tmp=paste('head(mappedkeys(',platform,'ENTREZID))',sep='')#eval(parse(text = tmp))###重点在这里,把字符串当做命令运行all_probe=eval(parse(text = paste('mappedkeys(',platform,'ENTREZID)',sep='')))EGID <- as.numeric(lookUp(all_probe, platformDB, "ENTREZID"))##自己把内容写出来即可}}
如果只是画网络图,那么只需要把所有的点,按照算好的坐标画出来,然后把所有的连线也画出即可!
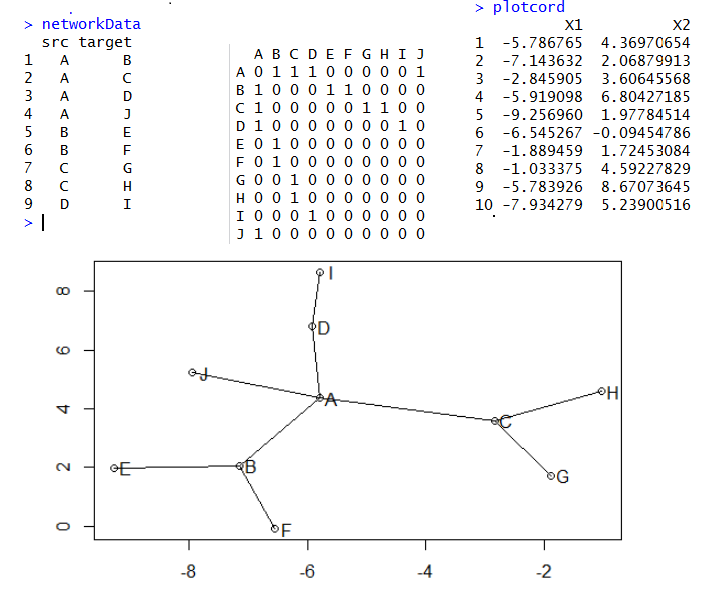

接下来, 我们直接看看R里面是如何画网络图的,我们首推一个包:networkD3/
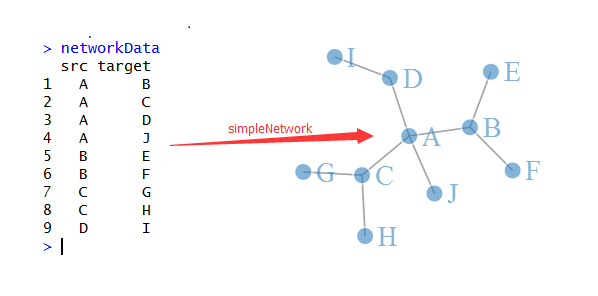
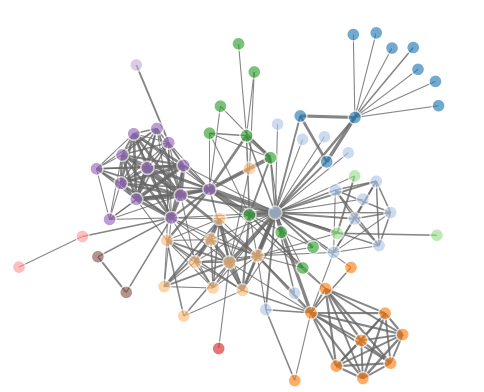
http://www.zhihu.com/question/19605599
> my_fun(10) [1] -2 2 2 0 [1] 2 3 1 1 [1] 0 4 2 1 [1] -4 5 3 1 [1] 4 6 1 2 [1] 6 7 1 3 [1] 8 8 1 4 [1] 6 9 2 4 [1] 2 10 3 4 [1] 10 11 1 5 [1] 8 12 2 5 [1] 8 如果我们赌11次,可以看到,我最后会剩余8块钱,每次输赢的情况都反应在里面了,可以自己模拟多次看看! 因为我只赌了11次,所以很快,如果我赌1000次,而且还想检验一下10000次模拟结果,就会比较慢了! 我首先使用进度条模拟一下结果,代码如下:还是比较慢的##Time difference of 1.861802 mins 我用了apply,好像时间是节省了一些,不过聊胜于无!
> library(pbapply) > start=Sys.time() > results=pbsapply(1:10000,function(i){my_fun(1000)}) |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% > Sys.time()-start ##9.909524 secs Time difference of 1.301539 mins 那接下来我们分析一下模拟的结果吧
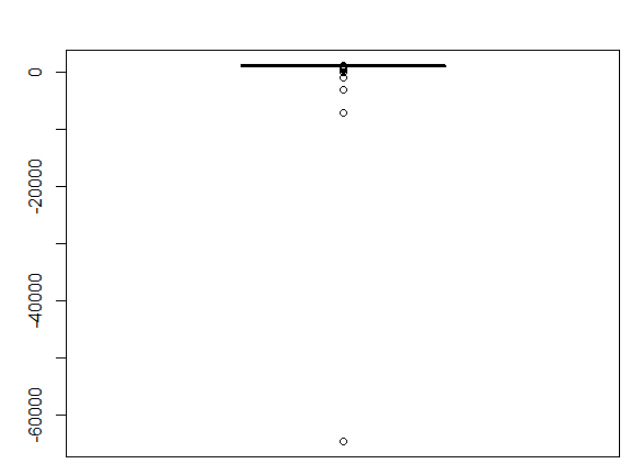
> summary(results) Min. 1st Qu. Median Mean 3rd Qu. Max. -64580 974 998 985 1020 1110 可以看到,我们平均下来是会赢钱的,而且赢面很大,赢的次数非常多!!! 但是,我们看下面的图就知道,一个很诡异的结果! 而且,我这里用的模拟胜负,并不是很好
这其实还只是小批量模拟,如果要模拟百亿次,首先我的笔记本肯定不行,cpu太破了,如果用服务器,就需要用并行计算啦! ##下面是用多核并行计算的代码,大家有兴趣可以自己去玩一下 library(parallel) cl.cores <- detectCores() cl <- makeCluster(cl.cores) clusterExport(cl, "judge") start=Sys.time() results=parSapply( cl=cl, 1:10000, my_fun(1000) ) Sys.time()-start ##4.260994 secs stopCluster(cl)
我们可以直接使用R的bioconductor里面的一个包,GOstats里面的函数来做超几何分布检验,看看每条pathway是否会富集
我们直接读取用limma包做好的差异分析结果
setwd("D:\\my_tutorial\\补\\用limma包对芯片数据做差异分析")
DEG=read.table("GSE63067.diffexp.NASH-normal.txt",stringsAsFactors = F)
View(DEG)
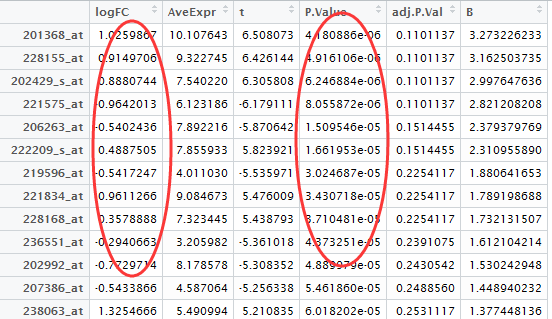
我们挑选logFC的绝对值大于0.5,并且P-value小雨0.05的基因作为差异基因,并且转换成entrezID
probeset=rownames(DEG[abs(DEG[,1])>0.5 & DEG[,4]<0.05,])
library(hgu133plus2.db)
library(annotate)
platformDB="hgu133plus2.db";
EGID <- as.numeric(lookUp(probeset, platformDB, "ENTREZID"))
length(unique(EGID))
#[1] 775
diff_gene_list <- unique(EGID)
这样我们的到来775个差异基因的一个list
首先我们直接使用R的bioconductor里面的一个包,GOstats里面的函数来做超几何分布检验,看看每条pathway是否会富集
library(GOstats)
library(org.Hs.eg.db)
#then do kegg pathway enrichment !
hyperG.params = new("KEGGHyperGParams", geneIds=diff_gene_list, universeGeneIds=NULL, annotation="org.Hs.eg.db",
categoryName="KEGG", pvalueCutoff=1, testDirection = "over")
KEGG.hyperG.results = hyperGTest(hyperG.params);
htmlReport(KEGG.hyperG.results, file="kegg.enrichment.html", summary.args=list("htmlLinks"=TRUE))
结果如下:
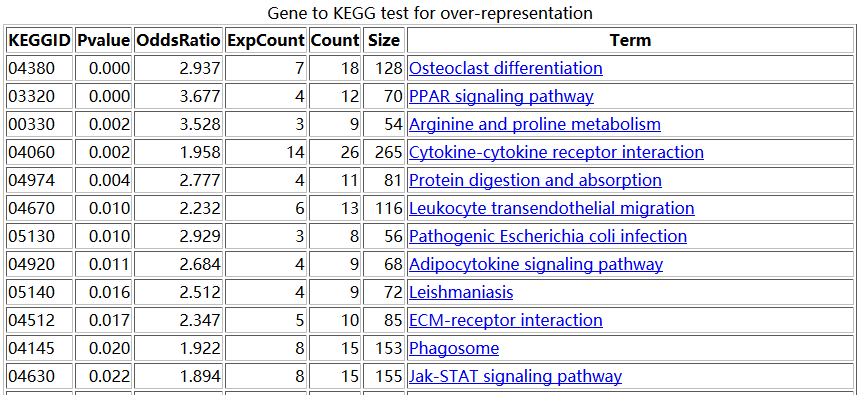
但是这样我们就忽略了其中的原理,我们不知道这些数据是如何算出来的,只是由别人写好的包得到了结果罢了。
事实上,这个包的这个hyperGTest函数无法就是包装了一个超几何分布检验而已。
如果我们了解了其中的统计学原理,我们完全可以写成一个自建的函数来实现同样的功能。
本来想着是如何习惯R里面的list对象,来实现hash的功能,无意中发现了居然还有这个包
https://cran.r-project.org/web/packages/hash/hash.pdf
我简单看了看文档,的确跟perl里面的hash功能类似,非常好用。
创建一个hash很简单
h <- hash( keys=letters, values=1:26 )
h <- hash( letters, 1:26 )
类似于下面的列表
L=setNames(as.list(LETTERS),1:26)
如果是列表要提取keys需要用names函数,如果需要提取values,需要用unlist函数,而用了hash之后,这两个函数就可以独自运行啦
还有很多其它函数,其实在list中也可以实现,主要是看你对哪种语法更熟悉,感觉自己现在编程能力差不多算是小有所成了,看什么都一样了,要是以前学R的时候看到了这个hash包,我肯定会很兴奋的!
clear signature(x = "hash"): Remove all key-value pairs from hash
del signature(x = "ANY", hash = "hash"): Remove specified key-value pairs from hash
has.key signature(key = "ANY", hash = "hash"): Test for existence of key
is.empty signature(x = "hash"): Test if no key-values are assigned
length signature(x = "hash"): Return number of key-value pairs from the hash
keys signature(hash = "hash"): Retrieve keys from hash
values signature(x = "hash"): Retrieve values from hash
copy signature(x = "hash"): Make a copy of a hash using a new environment.
format signature(x = "hash"): Internal function for displaying hash
AnnDbBimap对象是R里面的bioconductor系列包的基础对象,在探针数据里面会包装成ProbeAnnDbMap,跟go通路相关又是GOTermsAnnDbBimap对象。
但是它都是AnnDbBimap对象衍生过来的
主要存在于芯片系列的包和org系列的包,其实AnnDbBimap对象就是list对象的包装,比如下面这些例子:
ls("package:hgu133plus2.db")
| [1] "hgu133plus2" "hgu133plus2.db" "hgu133plus2_dbconn" |
| [4] "hgu133plus2_dbfile" "hgu133plus2_dbInfo" "hgu133plus2_dbschema" |
| [7] "hgu133plus2ACCNUM" "hgu133plus2ALIAS2PROBE" "hgu133plus2CHR" |
| [10] "hgu133plus2CHRLENGTHS" "hgu133plus2CHRLOC" "hgu133plus2CHRLOCEND" |
| [13] "hgu133plus2ENSEMBL" "hgu133plus2ENSEMBL2PROBE" "hgu133plus2ENTREZID" |
| [16] "hgu133plus2ENZYME" "hgu133plus2ENZYME2PROBE" "hgu133plus2GENENAME" |
| [19] "hgu133plus2GO" "hgu133plus2GO2ALLPROBES" "hgu133plus2GO2PROBE" |
| [22] "hgu133plus2MAP" "hgu133plus2MAPCOUNTS" "hgu133plus2OMIM" |
| [25] "hgu133plus2ORGANISM" "hgu133plus2ORGPKG" "hgu133plus2PATH" |
| [28] "hgu133plus2PATH2PROBE" "hgu133plus2PFAM" "hgu133plus2PMID" |
| [31] "hgu133plus2PMID2PROBE" "hgu133plus2PROSITE" "hgu133plus2REFSEQ" |
| [34] "hgu133plus2SYMBOL" "hgu133plus2UNIGENE" "hgu133plus2UNIPROT" |
那么我们可以随便挑选包中的一个数据分析一下
x <- hgu133plus2SYMBOL
xlist=as.list(x)
我们查看X对象,可知,它是object of class "ProbeAnnDbMap",而这个对象,就是常见的list对象,被包装了一下, 只有我们明白了它和list对象到底有什么区别,才算是真正搞懂了这一系列包
但是这个ProbeAnnDbMap对象,在GO等包里面会被包装成更复杂的对象-GOTermsAnnDbBimap,但是对他们的理解都大同小异。
我们先回顾一下在R语言里面的list的基础知识:
R 的 列表(list)是一个以对象的有序集合构成的对象。 列表中包含的对象又称为它的分量(components)。
分量可以是不同的模式或者类型, 如一个列表可以包括数值向量,逻辑向量,矩阵,复向量, 字符数组,函数等等
如果你会perl的话,可以把它理解成hash。
分量常常会被编号的(numbered),并且可以利用它来 访问分量
列表的分量可以被命名,这种情况下 可以通过名字访问。
特别要注意一下 Lst[[1]] 和 Lst[1] 的差别。 [[...]] 是用来选择单个 元素的操作符,而 [...] 是一个更为一般 的下标操作符。
因此前者得到的是列表 Lst 中的第一个对象, 并且含有分量名字的命名列表(named list)中分量的名字会被排除在外的。
后者得到的则是列表 Lst 中仅仅由第一个元素 构成的子列表。如果是命名列表, 分量名字会传给子列表的。
那么接下来我们就看看x和xlist的区别。
它们里面的数据都是affymetrix公司出品的人类的hgu133plus2芯片的探针ID与基因symbol的对应关系
如果我想拿到所有的探针ID,那么对于AnnDbBimap对象,需要用mappedkeys(x),对于普通的list对象,需要用names(xlist).
对于普通的list对象,如果我想看前几个元素,直接head就可以了,但是对于AnnDbBimap对象,数据被封装好了,需要先as.list,然后才能head
mapped_probes <- mappedkeys(x)
PID2=head(mapped_probes)
那么,如果我们想根据以下探针ID来查看它们在这些数据里面被对应着哪些基因symbol 呢?
> PID2 #这是一串探针ID,后面的操作都需要用的
[1] "1053_at" "117_at" "121_at" "1255_g_at" "1316_at" "1320_at"
如果是对于AnnDbBimap对象,我们可以用mget函数来操作,取多个探针的对应基因symbol
accnum <- mget(PID2, env=hgu133plus2ACCNUM);
gname <- mget(PID2, env=hgu133plus2GENENAME)
gsymbol <- mget(PID2, env=hgu133plus2SYMBOL)
mget函数返回的就是普通的list函数了,可以直接查看了。
如果是对于普通的list对象,我们想取多个探针的对应基因symbol也是非常简单的,xlist[PID2]即可。
那么我们不禁有问了,既然它们两个功能完全一样,何苦把它包装成一个对象了,我直接操作list对象不就好了,学那么多规矩干嘛?
所以,重点就来了
> length(mapped_probes)
[1] 42125
> length(names(xlist))
[1] 54675
看懂了吗?
但是,事实上用处也不大,你觉得下面这两个有区别吗?
SYMBOL <- AnnotationDbi::as.list(hgu133plus2SYMBOL)
SYMBOL <- SYMBOL[!is.na(SYMBOL)];
x <- hgu133plus2SYMBOL
mapped_probes <- mappedkeys(x)
SYMBOL <- AnnotationDbi::as.list(x[mapped_probes])
PS,在R里面创建一个list对象是非常简单的
The setNames() built-in function makes it easy to create a hash from given key and value lists.(Thanks to Nick K for the better suggestion.)
Usage: hh <- setNames(as.list(values), keys)
Example:
players <- c("bob", "tom", "tim", "tony", "tiny", "hubert", "herbert")
rankings <- c(0.2027, 0.2187, 0.0378, 0.3334, 0.0161, 0.0555, 0.1357)
league <- setNames(as.list(rankings), players)
经常会有人问这样的问题I have list of 10,000 Entrez IDs and i want to convert the multiple Entrez IDs into the respective gene names. Could someone suggest me the way to do this?等等类似的基因转换,能做的基因转换的方法非常多,以前不懂编程的时候,都是用各种网站,而最常用的就是ensembl的biomart了,它支持的ID非常多,高达几百种,好多ID我到现在都不知道是什么意思。
现在学会编程了,我比较喜欢的是R的一些包,是bioconductor系列,一般来说,其中有biomart,org.Hs.eg.db,annotate,等等。关于biomart我就不再讲了,我前面的博客至少有七八篇都提到了它。本次我们讲讲简单的, 我就以把gene entrez ID转换为gene symbol 为例子把。
当然,首先要安装这些包,并且加载。
if("org.Hs.eg.db" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("org.Hs.eg.db")}
suppressMessages(library(org.Hs.eg.db)) 我比较喜欢这样加载包
library(annotate) #一般都是这样加载包
如果是用org.Hs.eg.db包,首先你只需要读取你的待转换ID文件,构造成一个向量,tmp,然后只需要symbols <- org.Hs.egSYMBOL[as.character(tmp)]就可以得到结果了,返回的symbols是一个对象,需要用toTable这个函数变成数据框。但是这样转换容易出一些问题,比如如果你的输入数据tmp,里面含有一些无法转换的gene entrez ID,就会报错。
而且它支持的ID转换很有限,具体看看它的说明书即可:https://www.bioconductor.org/packages/release/data/annotation/manuals/org.Hs.eg.db/man/org.Hs.eg.db.pdf
org.Hs.eg.db
org.Hs.eg_dbconn
org.Hs.egACCNUM
org.Hs.egALIAS2EG
org.Hs.egCHR
org.Hs.egCHRLENGTHS
org.Hs.egCHRLOC
org.Hs.egENSEMBL
org.Hs.egENSEMBLPROT
org.Hs.egENSEMBLTRANS
org.Hs.egENZYME
org.Hs.egGENENAME
org.Hs.egGO
org.Hs.egMAP
org.Hs.egMAPCOUNTS
org.Hs.egOMIM
org.Hs.egORGANISM
org.Hs.egPATH
org.Hs.egPMID
org.Hs.egREFSEQ
org.Hs.egSYMBOL
org.Hs.egUCSCKG
org.Hs.egUNIGENE
org.Hs.egUNIPROT
如果是用annotate包,首先你还是需要读取你的待转换ID文件,构造成一个向量,tmp,然后用getSYMBOL(as.character(tmp), data='org.Hs.eg')这样直接就返回的还是以向量,只是在原来向量的基础上面加上了names属性。说明书:http://www.bioconductor.org/packages/3.3/bioc/manuals/annotate/man/annotate.pdf
然后你可以把转换好的向量写出去,如下:
1 A1BG
2 A2M
3 A2MP1
9 NAT1
10 NAT2
12 SERPINA3
13 AADAC
14 AAMP
15 AANAT
16 AARS
PS:如果是芯片数据,需要把探针的ID转换成gene,那么一般还需要加载特定芯片的数据包才行:
platformDB <- paste(eset.mas5@annotation, ".db", sep="") #这里需要确定你用的是什么芯片
cat("the annotation is ",platformDB,"\n")
if(platformDB %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");tmp=try(biocLite(platformDB))}
library(platformDB, character.only=TRUE)
probeset <- featureNames(eset.mas5)
rowMeans <- rowMeans(exprSet)
library(annotate) # lookUp函数是属于annotate这个包的
EGID <- as.numeric(lookUp(probeset, platformDB, "ENTREZID"))
理论上我前面提到的GEOquery包就可以根据一个GSE索引号来获取NCBI提供的所有关于这个GSE索引号的数据了,包括metadata,表达矩阵,soft文件,还有raw data
但是很多时候,那个metadata并不是很整齐,而且一个个下载太麻烦了,所以就需要用R的bioconductor的另一个神奇的包了GEOmetadb
它的示例:http://bioconductor.org/
library(GEOmetadb)
if(!file.exists('GEOmetadb.sqlite')) getSQLiteFile()
file.info('GEOmetadb.sqlite')
con <- dbConnect(SQLite(),'GEOmetadb.sqlite')
dbDisconnect(con)
但是一般不会成功,因为这个包把它的GEOmetadb.sqlite文件放在了国外网盘共享,在国内很难访问,推荐大家想办法下载到本地
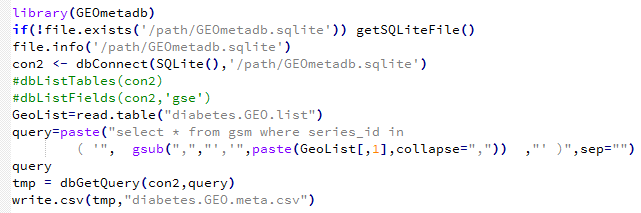 用这个代码就会成功了,需要自己下载GEOmetadb.sqlite文件然后放在指定目录:/path/GEOmetadb.sqlite 需要自己修改
我们的diabetes.GEO.list文件内容如下:
GSE1009
GSE10785
GSE1133
GSE11975
GSE121
GSE12409
那么会产生的表格文件如下:共有32列数据信息,算是蛮全面的了
用这个代码就会成功了,需要自己下载GEOmetadb.sqlite文件然后放在指定目录:/path/GEOmetadb.sqlite 需要自己修改
我们的diabetes.GEO.list文件内容如下:
GSE1009
GSE10785
GSE1133
GSE11975
GSE121
GSE12409
那么会产生的表格文件如下:共有32列数据信息,算是蛮全面的了
一直以来我都是随便看了点R的编程教程,因为我学了一点点C,所以还算有基础,现在基本上简单看看教程就能懂一门语言了,区别只是熟练度而已。R用得比较多,所以还算擅长,但是很多快捷应用的地方,我总是寄希望于到时候再查资料,所以没能用心的记住,这次花了点时间好好整理了一下R里面关于数据操作的重点,我想,以后再碰到类似的数据处理要求,应该很快能解决了把。
在R中,和排序相关的函数主要有三个:sort(),rank(),order()。
sort(x)是对向量x进行排序,返回值排序后的数值向量。
rank()是求秩的函数,它的返回值是这个向量中对应元素的“排名”。
order()的返回值是对应“排名”的元素所在向量中的位置。
其中sort(x)等同于x[order(x)]
下面以一小段R代码来举例说明:
> x<-c(97,93,85,74,32,100,99,67)
> sort(x)
[1] 32 67 74 85 93 97 99 100
> order(x)
[1] 5 8 4 3 2 1 7 6
> rank(x)
[1] 6 5 4 3 1 8 7 2
> x[order(x)]
[1] 32 67 74 85 93 97 99 100
其中比较有用的order,它可以用来给数据框进行排序
dat[order(dat[,1]),] 以该数据框的第一列进行排序
dat[order(dat[,1],dat[,2]),] 以该数据框的第一列为主要次序,第二列为次要序列进行排序
在R里面除了简单的对两个向量求交集并集补集之外,比较重要的就是match和 %in% 了,需要重点讲讲。
#首先对集合A,B,C赋值
> A<-1:10
> B<-seq(5,15,2)
> C<-1:5
> #求A和B的并集
> union(A,B)
[1] 1 2 3 4 5 6 7 8 9 10 11 13 15
> #求A和B的交集
> intersect(A,B)
[1] 5 7 9
> #求A-B
> setdiff(A,B)
[1] 1 2 3 4 6 8 10
> #求B-A
> setdiff(B,A)
[1] 11 13 15
> #检验集合A,B是否相同
> setequal(A,B)
[1] FALSE
> #检验元素12是否属于集合C
> is.element(12,C)
[1] FALSE
> #检验集合A是否包含C
> all(C%in%A)
[1] TRUE
> all(C%in%B)
从上面可以看到%in%这个操作符只返回逻辑向量TRUE 或者FALSE,而且返回值应该与%in%这个操作符前面的向量程度相等。也就是说它相当于遍历了C里面的一个个元素,判断它们是否在B中出现过,然后返回是或者否即可。
而match(C,B)的结果就很不一样了,它的返回结果同样与前面的向量等长,但是它并非返回逻辑向量,而是遍历了C里面的一个个元素,判断它们是否在B中出现过,如果出现就返回在B中的索引号,如果没有出现,就返回NA。
>B
[1] 5 7 9 11 13 15
>C
[1] 1 2 3 4 5
>match(C,B)
[1] NA NA NA NA 1
>C%in%B
[1] FALSE FALSE FALSE FALSE TRUE
这是一个需要安装的包,起得就是R里面最经典的把长型数据变宽,和把宽数据拉长的作用。
其中melt函数是把很宽的数据拉长,它就是需要指定几列数据是保证不被融合的, 其余每一列数据都必须被融合到一列了,融合后的这一列数据每个元素旁边就用列名来标记该数据来自于哪一列。
# example of melt function
library(reshape)
mdata <- melt(mydata, id=c("id","time"))
融合后的数据如下:
| id | time | variable | value |
| 1 | 1 | x1 | 5 |
| 1 | 2 | x1 | 3 |
| 2 | 1 | x1 | 6 |
| 2 | 2 | x1 | 2 |
| 1 | 1 | x2 | 6 |
| 1 | 2 | x2 | 5 |
| 2 | 1 | x2 | 1 |
| 2 | 2 | x2 | 4 |
可以看到variable列里面有两个不同的元素,说明是把旧数据中的两列给融合了,融合后的一个很长的列就是value
而cast函数的功能就是把刚才融合好的数据给还原。
# cast the melted data
# cast(data, formula, function)
subjmeans <- cast(mdata, id~variable, mean)
timemeans <- cast(mdata, time~variable, mean)
可以看到它们返回的结果是:
subjmeans
| id | x1 | x2 |
| 1 | 4 | 5.5 |
| 2 | 4 | 2.5 |
timemeans
| time | x1 | x2 |
| 1 | 5.5 | 3.5 |
| 2 | 2.5 | 4.5 |
可以看到cast函数比较复杂一点,formula公式右边的变量是需要拆开的variable,这一列变量有多少不重复元素,就新增多少列,左边的变量是行标记了,其余所有数据都会被计算一下再放在合适的位置。
在reshape2这个包里面还进化出来dcast和acast函数,功能大同小异。
这个函数的功能非常强大,类似于SQL语句里面的join系列函数
测试数据如下,它们这两个表的连接是作者名

如果要实现类似sql里面的inner join 功能,则用代码
m1 <- merge(authors, books, by.x = "surname", by.y = "name")
如果要实现left join功能则用代码
m1 <- merge(authors, books, by.x = "surname", by.y = "name",all.x=TRUE)
right join功能代码
m1 <- merge(authors, books, by.x = "surname", by.y = "name",all.y=TRUE)
all join功能代码
m1 <- merge(authors, books, by.x = "surname", by.y = "name",all=TRUE)
关于单变量匹配的总结就是这些,但对于多变量匹配呢,例如下面两个表,需要对k1,k2两个变量都相等的情况下匹配
x <- data.frame(k1 = c(NA,NA,3,4,5), k2 = c(1,NA,NA,4,5), data = 1:5)
y <- data.frame(k1 = c(NA,2,NA,4,5), k2 = c(NA,NA,3,4,5), data = 1:5)
匹配代码如下merge(x, y, by = c("k1","k2")) #inner join
另外一个多行匹配的例子如下:

我们的测试数据如上,这两个表的连接在于作者名。下面这两个语句等价
merge(authors, books, by=1:2)
merge(authors, books, by.x=c("FirstName", "LastName"),
by.y=c("AuthorFirstName", "AuthorLastName"),
all.x=TRUE)
都可以把两张表关联起来。
当然,在我搜索资料的时候,发现了另外一个解决问题的方法:
A[with(A, paste(C1, C2, sep = "\r")) %in% with(B, paste(C1, C2, sep="\r")), ]
参考:http://blog.sina.com.cn/s/blog_6caea8bf0100spe9.html
http://blog.sina.com.cn/s/blog_6caea8bf010159dt.html
http://blog.csdn.net/u014801157/article/details/24372441
http://www.statmethods.net/management/reshape.html
http://r.789695.n4.nabble.com/Matching-multiple-columns-in-a-data-frame-td890832.html
http://bbs.pinggu.org/thread-3234639-1-1.html
http://www.360doc.com/content/14/0821/13/18951041_403561544.shtml
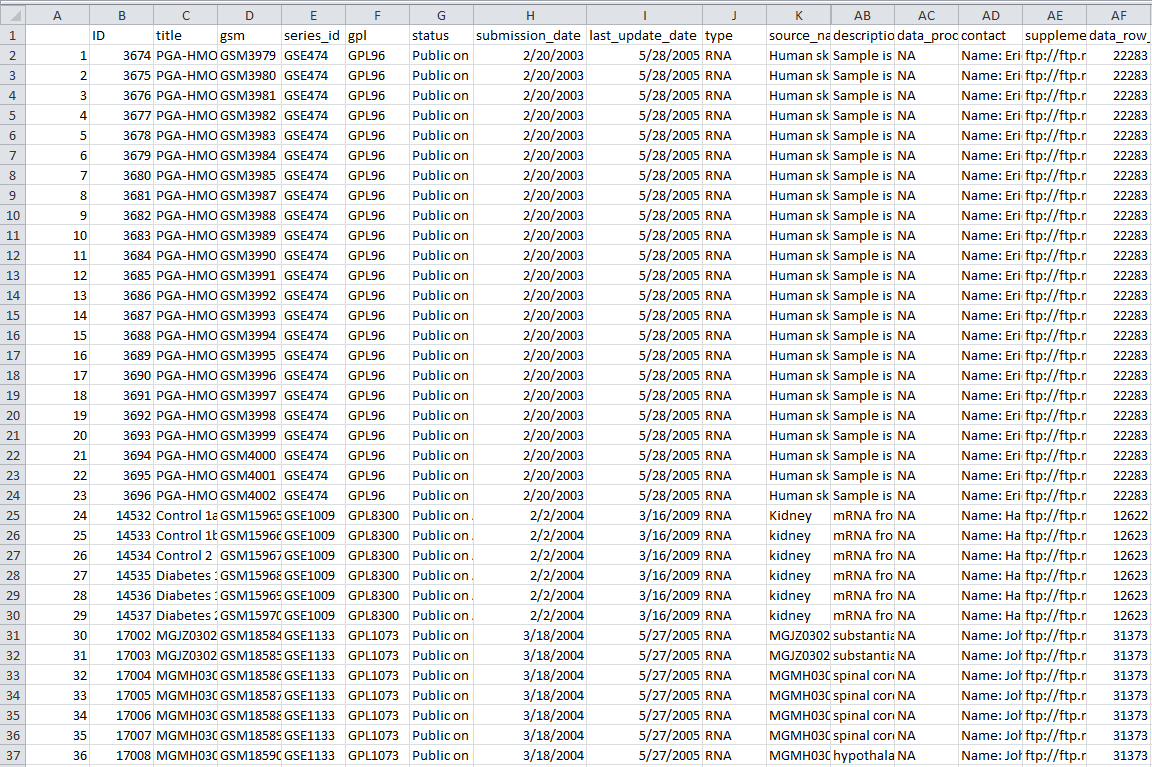
懂点芯片数据分析的都应该知道,芯片设计的时候,对一个基因设计了多个探针,这样设计是为了更好的捕获某些难以发现的基因,或者重点研究某些基因。
但是对我们的差异分析不方便,所以我们只分析哪些有对应了entrez ID的探针,而且对每个entrez ID,我们只需要挑选它表达量最高的那个探针。
所以就演化为一个编程问题:分组求最大值,多公共列合并!
如果是在R语言里面,那么首先这个table的表示形式如下
> esetDataTable[1:10,c(7,8)]
EGID rowMeans
1000_at 5595 1840.04259751826
1001_at 7075 799.075414422572
1002_f_at 1557 50.4884096416177
1003_s_at 643 142.372008051308
1004_at 643 211.65300963049
1005_at 1843 4281.29318032004
1006_at 4319 38.5784289213085
1007_s_at NA 1489.98158531843
1008_f_at 5610 4013.576753977
1009_at 3094 3070.50648167305
我们首先看看一个R语言函数处理方式吧,这个是比较容易想到的算法,但是用到了循环,非常的不经济,计算量很大。
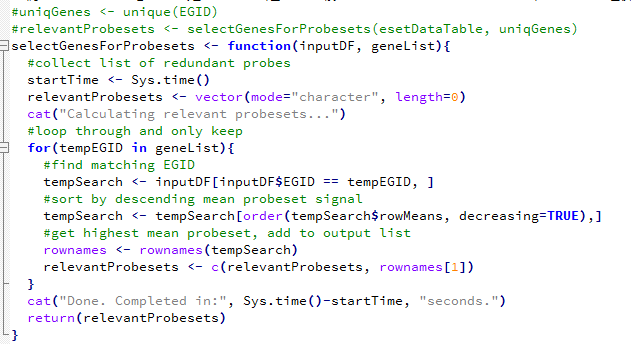
因为里面涉及到了双重循环。我进行了人工计时,这个程序耗费了一分钟十二秒,程序里面的计时器有点问题。
然后我再讲一个精简版的算法
dat=esetDataTable[,c(7,8)]
dat=as.data.frame(apply(dat,2,as.numeric))
dat$prob=rownames(esetDataTable)
首先可以得到需要提取的数据所在行的两个值
match_row=aggregate(dat,by=list(dat[,1]),max)
类似于这句SQL语句:SELECT max(a.rowMeans) as val, a.EGID FROM test.esetDataTable a group by a.EGID
现在要根据match_row表去原表esetDataTable里面提取我们的探针ID数据。

这样就完美解决了问题,我们的合并后的数据的prob列,就是前面那个函数计算了一分多钟的返回的非冗余探针ID向量,relevantProbesets,但是这次只花了不到一秒钟。
tmp_prob=merge(dat,match_row,by=c("EGID","rowMeans"))
setdiff(as.character(relevantProbesets),as.character(tmp_prob$prob))
length(union(as.character(relevantProbesets),as.character(tmp_prob$prob)))
setdiff(as.character(tmp_prob$prob),as.character(relevantProbesets))
我顺便检查了一下上面那个复杂的R函数跟我这次精简版的结果,发现这次才是对的,上面那个错了。
你们能发现上面那个为什么错了吗?
如果是在mysql里面它的表现形式如下;
mysql> select row_names,EGID,rowMeans from esetDataTable order by EGID limit 10;
+------------+-------+------------------+
| row_names | EGID | rowMeans |
+------------+-------+------------------+
| 38912_at | 10 | 85.8820246154773 |
| 41654_at | 100 | 301.720067595558 |
| 907_at | 100 | 273.100008206028 |
| 2053_at | 1000 | 354.207060199715 |
| 2054_g_at | 1000 | 33.8472900312781 |
| 40781_at | 10000 | 425.007640082848 |
| 35430_at | 10001 | 152.885791914329 |
| 35431_g_at | 10001 | 181.915087187117 |
| 35432_at | 10001 | 184.869017764782 |
| 31366_at | 10002 | 44.9716205901791 |
+------------+-------+------------------+
10 rows in set (0.05 sec)
如果要用SQL来做同样的事情需要下面这个语句,这个就非常简单啦!
select b.*
from test.esetDataTable b
inner join (
SELECT max(a.rowMeans) as val, a.EGID FROM test.esetDataTable a group by a.EGID
) as c on b.EGID=c.EGID and b.rowMeans=c.val
结果是:8640 rows in set (4.56 sec) 看起来mysql还没有R语言快速,但是这个mysql语句很明显也不是很高效,但是我的mysql水平有限。
稍微解释一下这个mysql语句,其中SELECT max(a.rowMeans) as val, a.EGID FROM test.esetDataTable a group by a.EGID类似于R里面的match_row=aggregate(dat,by=list(dat[,1]),max),就是把每个entrez ID对应着的最大值提取出来成一个新的表
而inner join on b.EGID=c.EGID and b.rowMeans=c.val 就是我们的tmp_prob=merge(dat,match_row,by=c("EGID","rowMeans")) 根据两列来合并两个数据框