mutation signature这个概念提出来还不久,我看了看文献,最早见于2013年的一篇nature文章,主要是用来描述癌症患者的somatic mutation情况的。
首先要自己分析癌症样本数据,拿到somatic mutation,TCGA计划发展到现在已经有非常多的somatic mutation结果啦,大家可以自行选择感兴趣的癌症数据拿来研究,解析一下mutation signature 。
我这里给大家推荐一个工具,是R语言的Bioconductor系列包中的一个,SomaticSignatures
其实它的说明书写的非常详细了已经,如果你理解了mutation signature的概念,很容易用那个包,其实你自己写一个脚本也是非常任意的,就是根据mutation的位置在基因组中找到它的前后一个碱基,然后组成三碱基突变模式,最后统计一下那96种突变模式的分布状况!
我这里简单讲一讲这个包如何用吧!
首先下载并加载几个必须的包:
library(SomaticSignatures) ## 程序
library(SomaticCancerAlterations) ## 自带测试数据
library(BSgenome.Hsapiens.1000genomes.hs37d5) ## 我们的参考基因组
library(VariantAnnotation)
## 这个对象很重要: GRanges class of the GenomicRanges package
##其中SomaticCancerAlterations这个包提供了测试数据,来自于8个不同癌症的外显子测序的项目。
sca_metadata = scaMetadata()
###可以查看关于这8个项目的介绍,每个项目都测了好几百个样本。但是我们只关心突变数据,而且只关心somatic的突变数据。
sca_data = unlist(scaLoadDatasets())
然后根据突变数据做好一个GRanges对象,这个可以看我以前的博客
sca_data$study = factor(gsub("(.*)_(.*)", "\\1", toupper(names(sca_data))))
sca_data = unname(subset(sca_data, Variant_Type %in% "SNP"))
sca_data = keepSeqlevels(sca_data, hsAutosomes())
## 这个对象就是我们软件的输入数据
sca_vr = VRanges(
seqnames = seqnames(sca_data),
ranges = ranges(sca_data),
ref = sca_data$Reference_Allele,
alt = sca_data$Tumor_Seq_Allele2,
sampleNames = sca_data$Patient_ID,
seqinfo = seqinfo(sca_data),
study = sca_data$study
)
## 这里还可以直接用readVcf或者readMutect 来读取本地somatic mutation文件
## 提取突变数据,并且构造成一个Range对象。
sca_vr
###可以简单看看每个study都有多少somatic mutation
sort(table(sca_vr$study), decreasing = TRUE)
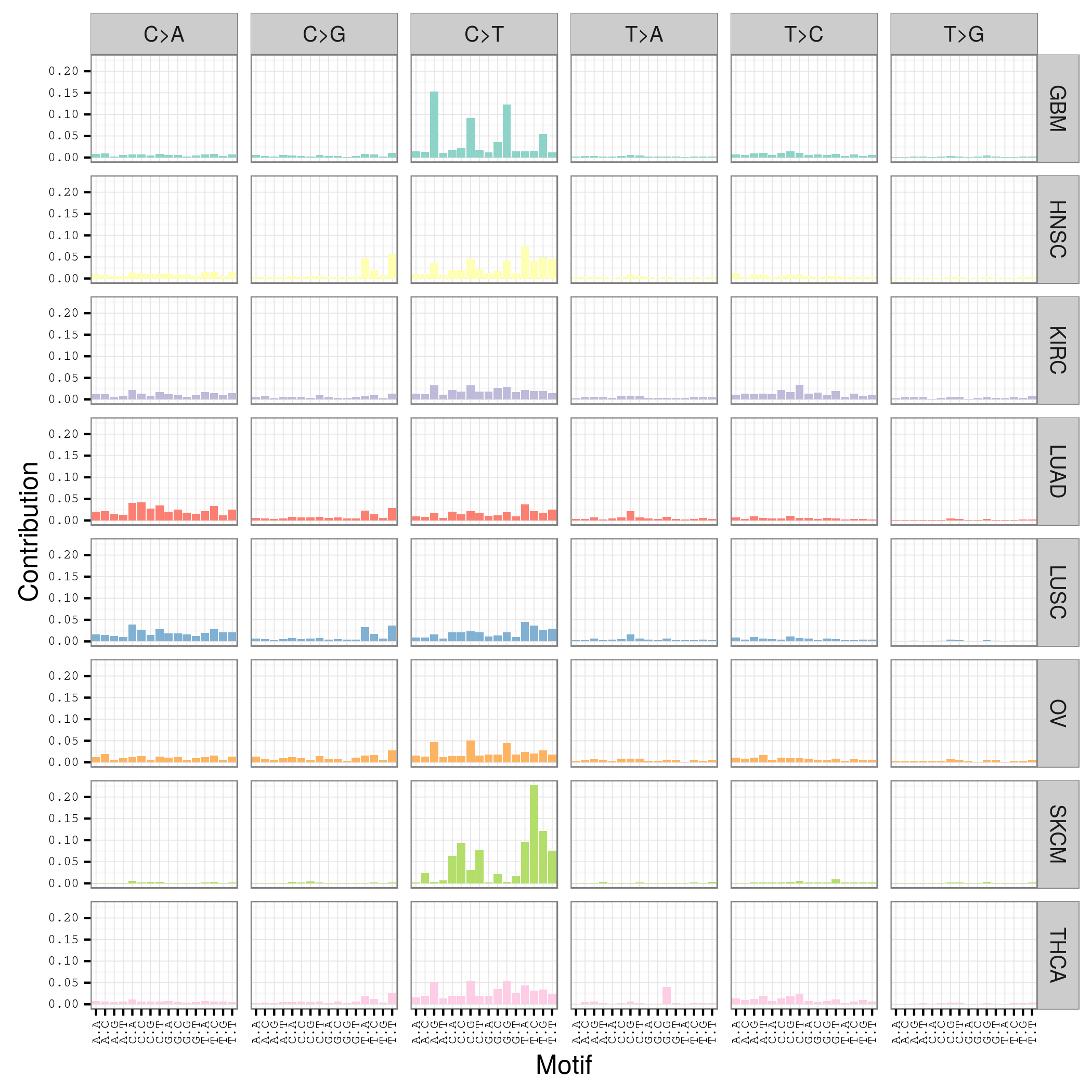
LUAD SKCM HNSC LUSC KIRC GBM THCA OV
208724 200589 67125 61485 24158 19938 6716 5872
##用mutationContext函数来根据Range对象和下载好的参考基因组文件来获取突变的上下文信息。
sca_motifs = mutationContext(sca_vr, BSgenome.Hsapiens.1000genomes.hs37d5)
head(sca_motifs)
##可以看到Range对象,增加了两列:alteration context
## 接下来根据做好的上下文突变数据矩阵来构建 the matrix MM of the form {motifs × studies}
sca_mm = motifMatrix(sca_motifs, group = "study", normalize = TRUE)
## 根据96种突变的频率,而不是次数来构造矩阵
head(round(sca_mm, 4))
## 然后直接画出每个study的Mutation spectrum 图
plotMutationSpectrum(sca_motifs, "study")
## 还要把spectrum分解成signature!!
## 这个包提供了两种方法,分别是NMF和PCA
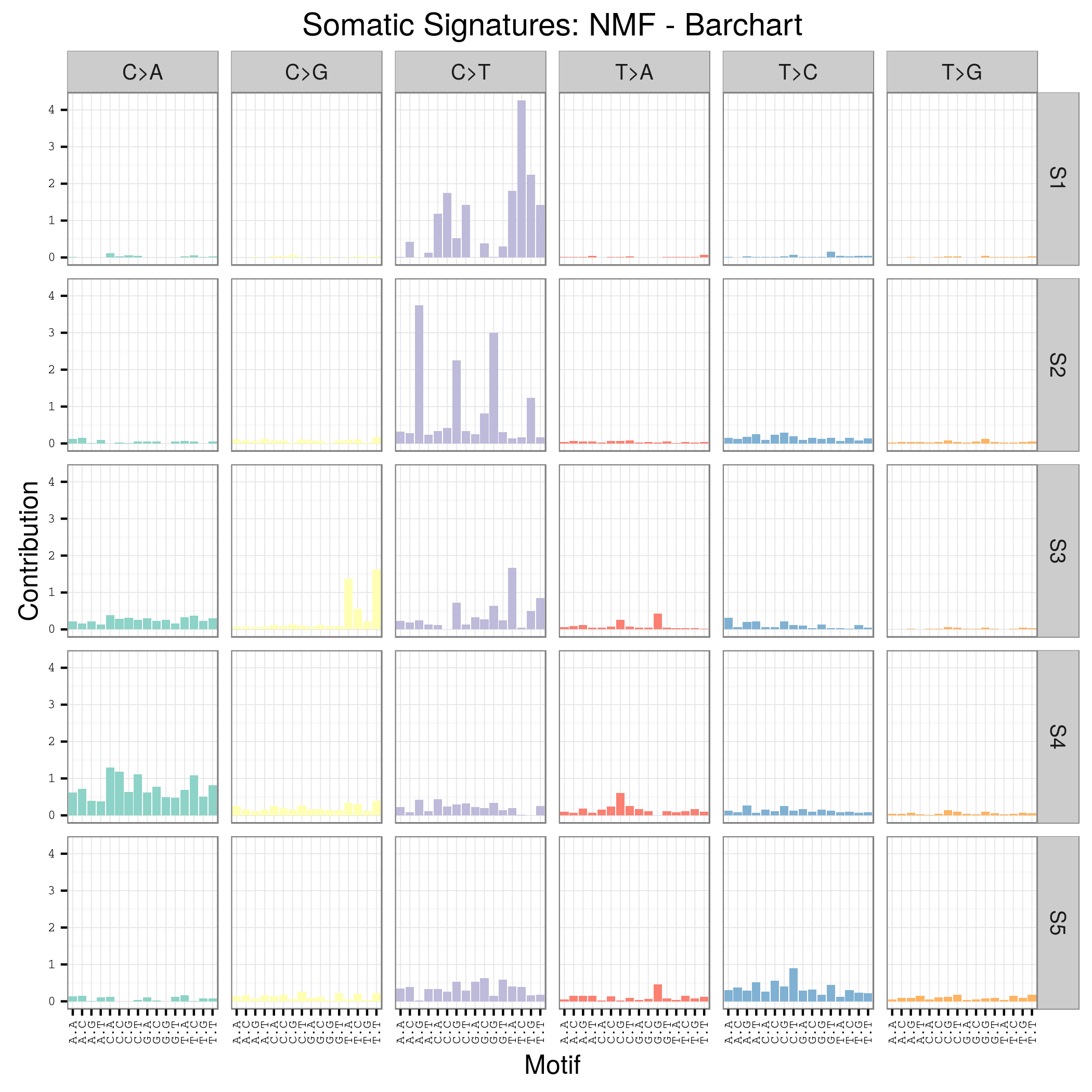
n_sigs = 5
sigs_nmf = identifySignatures(sca_mm, n_sigs, nmfDecomposition)
sigs_pca = identifySignatures(sca_mm, n_sigs, pcaDecomposition)
##还提供了很多函数来探索:signatures, samples, observed and fitted.
需要我们掌握的是assessNumberSignatures,用来探索我们到底应该把spectrum分成多少个signature
n_sigs = 2:8
gof_nmf = assessNumberSignatures(sca_mm, n_sigs, nReplicates = 5)
gof_pca = assessNumberSignatures(sca_mm, n_sigs, pcaDecomposition)
plotNumberSignatures(gof_nmf) ## 可视化展现
## 接下来可视化展现具体每个cancer type里面的各个个体在各个signature的占比
library(ggplot2)
plotSignatureMap(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Heatmap")
plotSignatures(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Barchart")
plotObservedSpectrum(sigs_nmf)
plotFittedSpectrum(sigs_nmf)
plotSampleMap(sigs_nmf)
plotSamples(sigs_nmf)
同理,PCA的结果也可以同样的可视化展现:
plotSignatureMap(sigs_pca) + ggtitle("Somatic Signatures: PCA - Heatmap")
plotSignatures(sigs_pca) + ggtitle("Somatic Signatures: PCA - Barchart")
plotFittedSpectrum(sigs_pca)
plotObservedSpectrum(sigs_pca)
值得一提的是,所有的plot系列函数,都是基于ggplot的,所以可以继续深度定制化绘图细节。
p = plotSamples(sigs_nmf)
## (re)move the legend
p = p + theme(legend.position = "none")
## (re)label the axis
p = p + xlab("Studies")
## add a title
p = p + ggtitle("Somatic Signatures in TGCA WES Data")
## change the color scale
p = p + scale_fill_brewer(palette = "Blues")
## decrease the size of x-axis labels
p = p + theme(axis.text.x = element_text(size = 9))
###当然,对上下文突变数据矩阵也可以进行聚类分析
clu_motif = clusterSpectrum(sca_mm, "motif")
library(ggdendro)
p = ggdendrogram(clu_motif, rotate = TRUE)
p
## 最后,由于我们综合了8个不同的study,所以必然会有批次影响,如果可以,也需要去除。