我以前写过DESeq,以及过时了:http://www.bio-info-trainee.com/867.html
正好准备筹集bioconductor中文社区,我写简单讲一下DESeq2这个包如何用!
我以前写过DESeq,以及过时了:http://www.bio-info-trainee.com/867.html
正好准备筹集bioconductor中文社区,我写简单讲一下DESeq2这个包如何用!
开发单位:华大,SOAP系列软件套装!
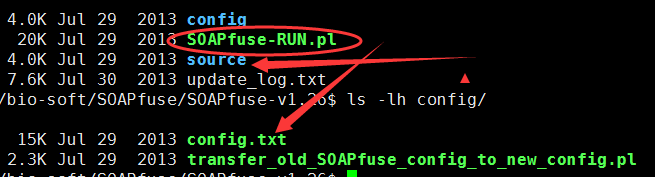
二,输入数据准备
6.5K Jun 15 2009 cytoBand.txt.gz3.0G Oct 12 2012 hg19.fa2.5M Mar 15 10:30 HGNC_Gene_Family_dataset38M Feb 8 2014 Homo_sapiens.GRCh37.75.gtf.gz202 Jan 19 16:07 HumanRef_refseg_symbols_relationship.list
文件下载地址,作者已经给出了!
这一步耗时很长,4~6小时,创造了transcript.fa和gene.fa,然后还对他们建立bwa和soap的index,所以有点慢!
Congratulations!You have constructed SOAPfuse database files successfully.These database files are all stored in directory you supplied:/home/jmzeng/biosoft/SOAPfuse/db_v1.27/They are all generated based on public data files you supplied:whole_genome_fasta_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/hg19.fagtf_annotation_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/Homo_sapiens.GRCh37.75.gtf.gzChr_Bandregion_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/cytoBand.txt.gzHGNC_gene_family_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/HGNC_Gene_Family_datasetgtf_segname2refseg_list: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/HumanRef_refseg_symbols_relationship.list
DB_db_dir = /DATABASE_DIR/PG_pg_dir = /TOOL_DIR/source/binPS_ps_dir = /TOOL_DIR/sourcePD_all_out = /out_directory/PA_all_fq_postfix = PostFix
三,运行命令
perl SOAPfuse-RUN.pl -c <config_file> -fd <WHOLE_SEQ-DATA_DIR> -l <sample_list> -o <out_directory> [Options]
运行的非常慢!!!
四,数据结果解读
This online tool currently uses CrossMap, which supports a limited number of formats (see our online documentation for details of the individual data formats listed below). CrossMap also discards metadata in files, so track definitions, etc, will be lost on conversion.
Important note: CrossMap converts WIG files to BedGraph internally for efficiency, and also outputs them in BedGraph format.
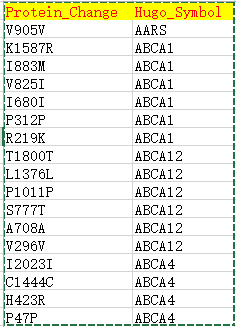
| Hugo_Symbol | HUGO symbol for the gene | TP53 |
| Protein_Change | Amino acid change | V600E |
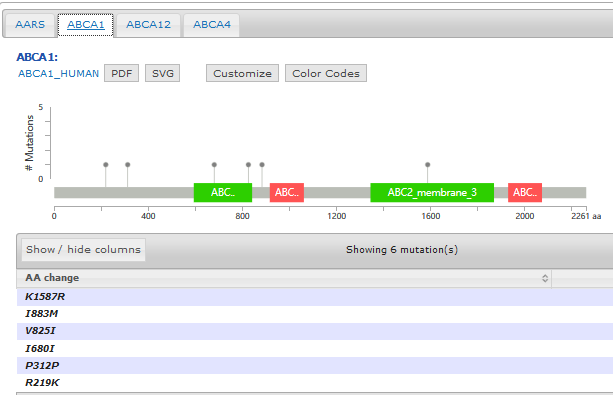
Pfam provides an online tool to not only generate the domain information in JSON format, but to draw the lollipop diagram using javascript as well. They have more information here: http://pfam.xfam.org/help#tabview=tab9
IMHO, not as pretty as cBioPortal's but it gets you close to a solution.
EDIT / SHAMELESS PLUG: After seeing the data available and how easy it'd be, I made my own quick tool to fetch the data and draw the diagram for me in a style similar to cBioPortal - feel free to fork it and add features: https://github.com/pbnjay/lollipops
Example output (w/ labels per the comments)
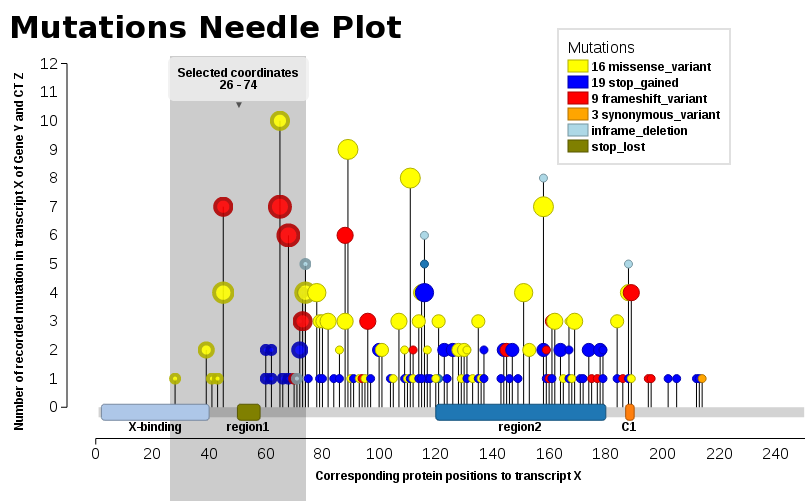
We found ourselves in the same need, we wanted such a plot (JavaScript). Thus, I add our solution, Mutations Needle Plot. The library creates an SVG image (with D3), which then may be downloaded.
You will npm in order to be able to install & run the library.
Examples may be found in the snippets folder or also the index.html - The one displayed here below
我现在还不是很确定这个方法,只是试一试,欢迎与我交流对该方法的讨论!
Wang H, Sun Q, Zhao W, et al. Individual-level analysis of differential expression of genes and pathways for personalized medicine[J]. Bioinformatics, 2014: btu522.
他们把它写成了一个R包,可以下载使用,但是必须用R2.15.2版本,我用了一下,不好用!
We can download the R code for in http://bioinformatics.oxfordjournals.org/content/31/1/62/suppl/DC1
他们这个程序真心不好用,但是很容易看懂算法,可以自己用R语言写一个来实现同样的过程!
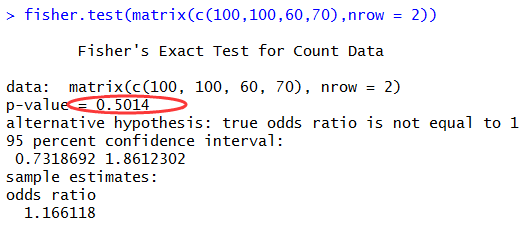
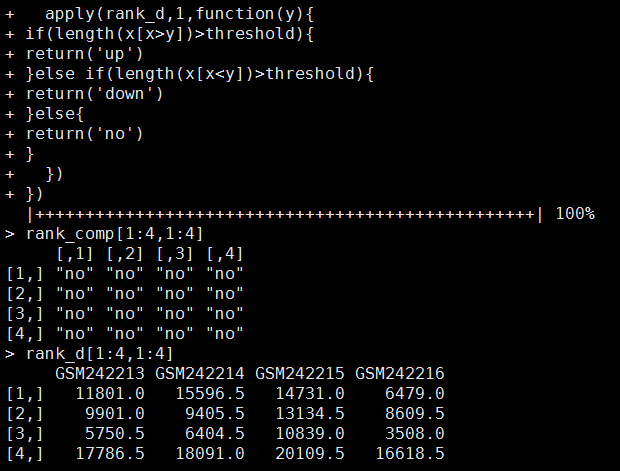
报错如下:ERROR MESSAGE: SAM/BAM file input.marked.bam is malformed: SAM file doesn't have any read groups defined in the header. The GATK no longer supports SAM files without read groups !

之前用过有界面的那种,那样非常方便,只需要做好数据即可,但是如果有非常多的数据,每次都要点击文件,点击下一步,也很烦,不过,,它既然是java软件,就可以以命令行的形式来玩转它!
直接在官网下载java版本软件即可:http://software.broadinstitute.org/gsea/downloads.jsp
需要下载gmt文件,自己制作gct和cls文件,或者直接下载测试文件p53
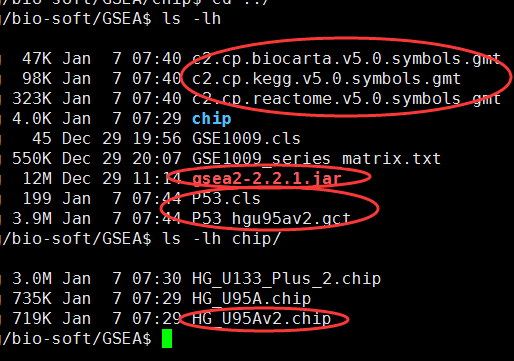
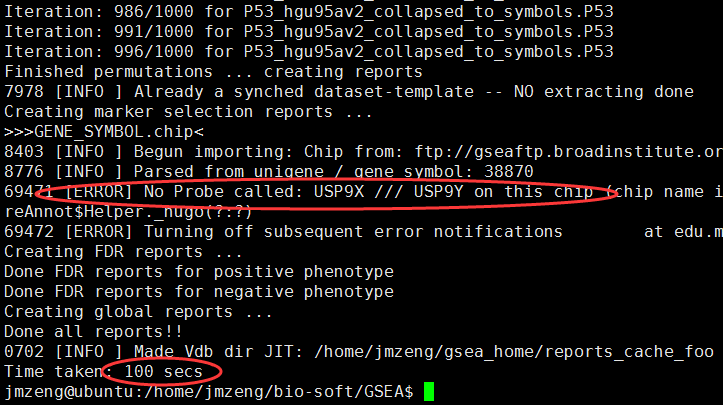
hgu95av2的芯片数据,只有一万多探针,所以很快就可以出结果
| CP: Canonical pathways (browse 1330 gene sets) |
Gene sets from the pathway databases. Usually, these gene sets are canonical representations of a biological process compiled by domain experts. details | Download GMT Files original identifiers gene symbols entrez genes ids |
|---|---|---|
| CP:BIOCARTA: BioCarta gene sets (browse 217 gene sets) |
Gene sets derived from the BioCarta pathway database (http://www.biocarta.com/genes/index.asp). | Download GMT Files original identifiers gene symbols entrez genes ids |
| CP:KEGG: KEGG gene sets (browse 186 gene sets) |
Gene sets derived from the KEGG pathway database (http://www.genome.jp/kegg/pathway.html). | Download GMT Files original identifiers gene symbols entrez genes ids |
| CP:REACTOME: Reactome gene sets (browse 674 gene sets) |
Gene sets derived from the Reactome pathway database (http://www.reactome.org/). | Download GMT Files original identifiers gene symbols entrez genes ids |
然后我看了一下域名,发现其实很有规律的
![[DIR]](file:///C:/Users/jmzeng/AppData/Local/YNote/data/jmzeng1314@163.com/901f79782af94aecb22c22dcb42c10a4/folder.gif) |
linux/ | 23-Jan-2008 19:47 | - | |
|
macos/ | 19-Apr-2005 09:45 | - | |
|
macosx/ | 12-Dec-2015 09:04 | - | |
|
windows/ | 24-Feb-2012 18:41 | - |
|
debian/ | 15-Dec-2015 02:06 | - | |
|
redhat/ | 27-Jul-2014 21:12 | - | |
|
suse/ | 16-Feb-2012 15:09 | - | |
|
ubuntu/ | 06-Jan-2016 04:05 | - |
|
precise/ | 06-Jan-2016 04:03 | - | |
|
trusty/ | 06-Jan-2016 04:04 | - | |
|
vivid/ | 06-Jan-2016 04:04 | - | |
|
wily/ | 06-Jan-2016 04:05 | - |
所以如果,大家是想在linux系统里面安装旧版本的R,建议大家直接下载c源码,直接三部曲就可以安装啦!
这里的拷贝数变异检测芯片指的是Affymetrix Genome-Wide Human SNP Array 6.0
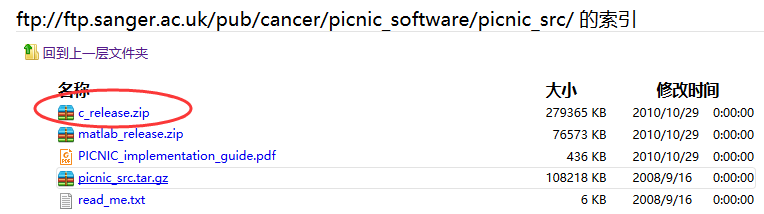
jmzeng@ubuntu:/home/jmzeng/bio-soft/picnic/c_code/celConverter$ java -jar CelFileConverter.jar -m Snp6FeatureMappings.csv -c ../cdf/GenomeWideSNP_6.Full.cdf -s ../celFiles/ -t ../result/
Loading CDF file: ../cdf/GenomeWideSNP_6.Full.cdf
Loading feature mapping file: Snp6FeatureMappings.csv
Processing CEL file: /home/jmzeng/bio-soft/picnic/c_code/celConverter/../celFiles/GSM1949207.CEL
Created feature intensity file: /home/jmzeng/bio-soft/picnic/c_code/celConverter/../result/GSM1949207.feature_intensity
Processing CEL file: /home/jmzeng/bio-soft/picnic/c_code/celConverter/../celFiles/GSM887898.CEL
Created feature intensity file: /home/jmzeng/bio-soft/picnic/c_code/celConverter/../result/GSM887898.feature_intensity

HMM('GSM1949207.feature_intensity','info\','result\output\','result\',10,0,2.0221,0.40997)

ACC BLCA BRCA CESC COAD COADREAD DLBC ESCA GBM HNSC KICH KIRC KIRP LAML LGG LIHC LUAD LUSC OV PAAD PANCANCER PANCAN8 PANCAN12 PRAD READ SARC SKCM STAD THCA UCEC UCS
官网:http://mutationassessor.org/
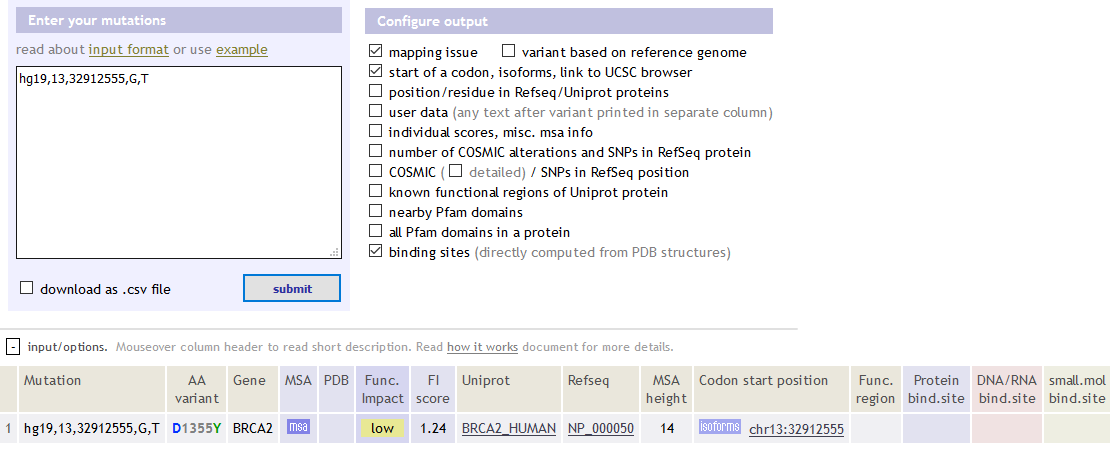
hg19,13,32912555,G,T BRCA2
hg18,7,55178574,G,A GBM
7,55178574,G,A GBM
or in protein space: <protein ID> <variant> <text>, where <protein ID> can be :
1. Uniprot protein accession (i.e. EGFR_HUMAN)
2. NCBI Refseq protein ID (i.e. NP_005219)
examples:
EGFR_HUMAN R521K
EGFR_HUMAN R98Q Polymorphism
EGFR_HUMAN G719D disease
NP_000537 G356A
NP_000537 G360A dbSNP:rs35993958
NP_000537 S46A Abolishes phosphorylation
ID types can be mixed in one list in any way.
其实主要要讲的不是用excel来做差异分析,只是想讲清楚差异分析的原理,用excel可视化的操作可能会更方便理解,而且想告诉大家,其实生物信息学分析,本来就很简单的,那么多软件,只有你理解了原理,你自己就能写出来的!
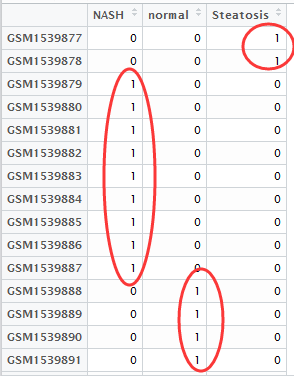
首先,还是得到表达矩阵,下面绿色的样本是NASH组,蓝色的样本是normal组
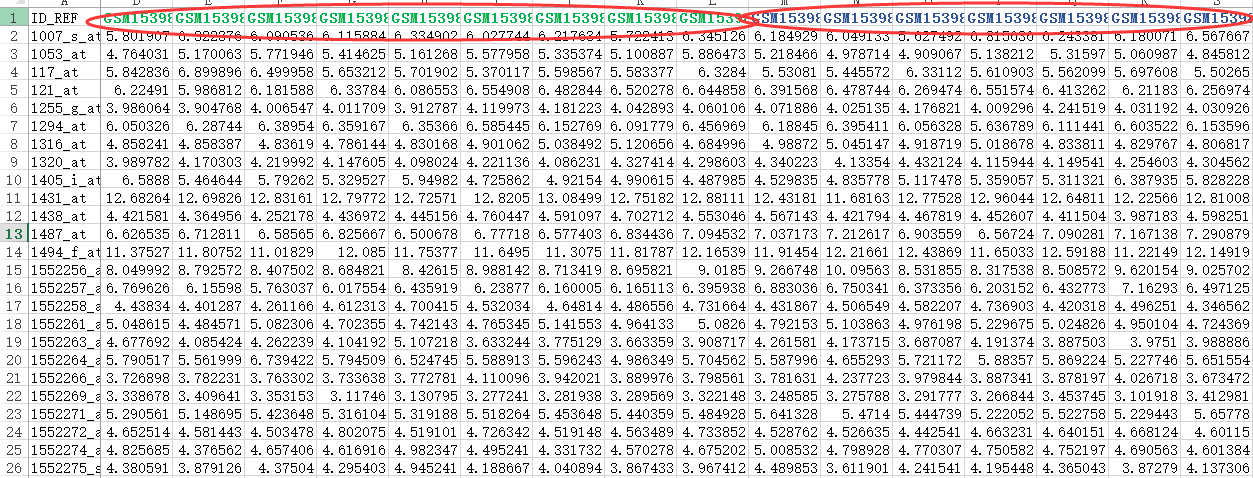
我们进行差异分析,很简单,就是看两组的表达值,是否差异,而检验的方法就是T检验。
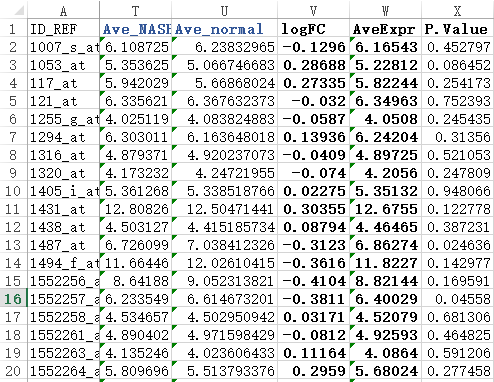
=AVERAGE(D2:L2) ##求NASH组的平均表达量
=AVERAGE(M2:S2) ###求normal的平均表达量
=T2-U2 ##计算得到logFOLDchange值
=AVERAGE(D2:S2) ###得到所有样本的平均表达量
=T.TEST(D2:L2,M2:T2,2,3) ###用T检验得到两个组的表达量的差异显著程度。
简单检查几个值就可以看到跟limma包得到的结果差不多。
下载该R语言包,然后看说明书,需要自己做好三个数据(表达矩阵,分组矩阵,差异比较矩阵),总共三个步骤(lmFit,eBayes,topTable)就可以啦

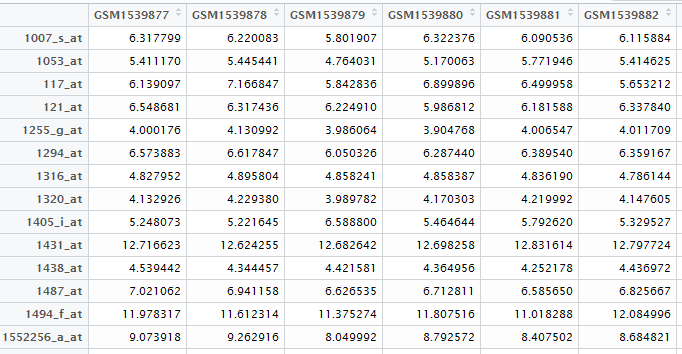
首先做第一个数据,基因表达矩阵!
自己在NCBI里面可以查到下载地址,然后用R语言读取即可
exprSet=read.table("GSE63067_series_matrix.txt.gz",comment.char = "!",stringsAsFactors=F,header=T)
rownames(exprSet)=exprSet[,1]
exprSet=exprSet[,-1]
然后做好分组矩阵,如下
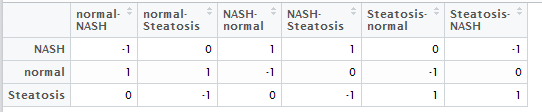
然后做好,差异比较矩阵,就是说明你想把那些组拿起来做差异分析,如下
最后输出结果:
我进行了6次比较,所以会输出6次比较结果
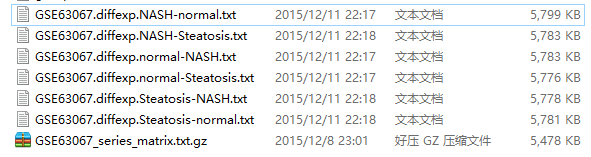
最后打开差异结果,解读,说明书如下!
在我的github有完整代码
matlab毕竟是收费软件,而且是有界面的。所以搞生物信息的都用R和linux替代了,但是很多高大上的单位,比如大名鼎鼎的broadinstitute,是用matlab的,所以他们开发的程序也会以matlab代码的形式发布。但是考虑到大多研究者用不起matlab,或者不会用,所以就用linux系统里面安装matlab运行环境来解决这个问题,我们仍然可以把人家写的matlab程序,在linux命令行下面,当做一个脚本来运行!
比如,这次我就需要用broadinstitute的一个软件:Mutsig,找cancer driver gene的,http://www.broadinstitute.org/cancer/cga/mutsig_run,但是我看了说明才发现,它是用matlab写的,所以我要想在我的服务器用,就必须按照安装matlab运行环境,在官网可以下载:http://www.mathworks.com/products/compiler/mcr/
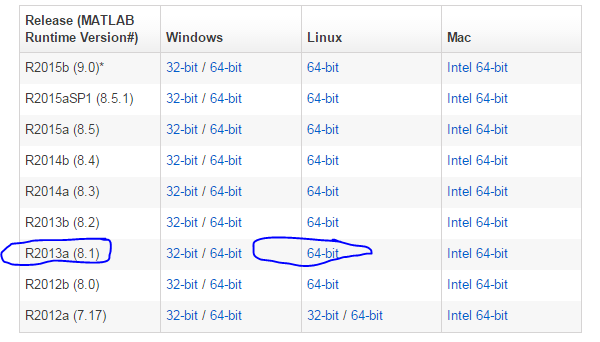
我这里选择的是R2013a (8.1),下载之后解压是这样的,压缩包约四百多M
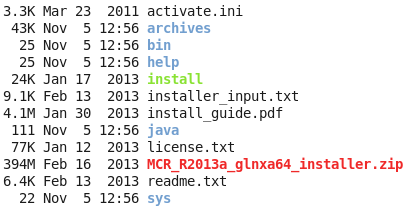
然后直接在解压后的目录里面运行那个install即可,然后如果你的linux可以传送图像,那么就会想安装windows软件一样方便!如果你的linux是纯粹的命令行,那么,就需要一步步的命令行交互,选择安装地址,等等来安装了。
记住你安装之后,会显示一些环境变量给你,请千万要记住,然后自己去修改自己的环境变量,如果你忘记了,就需要搜索来解决环境变量的问题啦!安装之后是这样的:
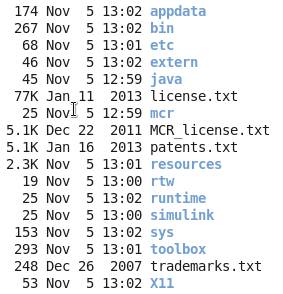
请记住你的安装目录,以后你运行其它matlab相关的程序,都需要把这个安装目录,当做一个参数传给你的其它程序的!!!
如果你没有设置环境变量,就会出各种各样的错误,用下面这个脚本可以设置
其中MCRROOT一般是$path/biosoft/matlab_running/v81/ 这样的东西,请务必注意,LD_LIBRARY_PATH非常重要,非常重要,非常重要!!!!
MCRROOT=$1
echo ---
LD_LIBRARY_PATH=.:${MCRROOT}/runtime/glnxa64 ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRROOT}/bin/glnxa64 ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRROOT}/sys/os/glnxa64;
MCRJRE=${MCRROOT}/sys/java/jre/glnxa64/jre/lib/amd64 ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE}/native_threads ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE}/server ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE}/client ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE} ;
XAPPLRESDIR=${MCRROOT}/X11/app-defaults ;
export LD_LIBRARY_PATH;
export XAPPLRESDIR;
echo LD_LIBRARY_PATH is ${LD_LIBRARY_PATH};
如果你没有设置正确,那么会报一下的错误!
error while loading shared libraries: libmwmclmcrrt.so.8.1: cannot o pen shared object file: No such file or directory
error while loading shared libraries: libmwlaunchermain.so: cannot o pen shared object file: No such file or directory
等等!!!
wget http://clavius.bc.edu/~erik/freebayes/freebayes-5d5b8ac0.tar.gz
tar xzvf freebayes-5d5b8ac0.tar.gz
cd freebayes
make
一个小插曲,安装的过程报错:/bin/sh: 1: cmake: not found
所以我需要自己下载安装cmake,然后把cmake添加到环境变量
首先下载源码包http://www.cmake.org/cmake/resources/software.html
wget http://cmake.org/files/v3.3/cmake-3.3.2.tar.gz
解压进去,然后源码安装三部曲,首先 ./configu 然后make 最后make install
cmake 会默认安装在 /usr/local/bin 下面,这里需要修改,因为你可能没有 /usr/local/bin 权限,安装到自己的目录,然后把它添加到环境变量!
step2:准备数据
wget http://bioinformatics.bc.edu/marthlab/download/gkno-cshl-2013/chr20.fawget ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/data/NA12878/alignment/NA12878.chrom20.ILLUMINA.bwa.CEU.low_coverage.20121211.bam
wget ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/data/NA12878/alignment/NA12878.chrom20.ILLUMINA.bwa.CEU.low_coverage.20121211.bam.bai用这个代码就可以下载千人基因组计划的NA12878样本的第20号染色体相关数据啦
freebayes -f chr20.fa \
NA12878.chrom20.ILLUMINA.bwa.CEU.low_coverage.20121211.bam >NA12878.chr20.freebayes.vcf一般就用默认参数即可
基于高通量测序数据进行HLA分型的软件挺多的,比较老的有三个,作者分别是Boegel et al.Kim et al.Major et al.,然后他们都被OptiType这个软件的作者被批评了,我这里先介绍Kim et al的seq2HLA使用方法,以下是它的一些链接。
功能概述
seq2HLA is a computational tool to determine Human Leukocyte Antigen (HLA) directly from existing and future short RNA-Seq reads. It takes standard RNA-Seq sequence reads in fastq format as input, uses a bowtie index comprising known HLA alleles and outputs the most likely HLA class I and class II types, a p-value for each call, and the expression of each class.
软件简介
Type of tool Program
Nature of tool Standalone
Operating system Unix/Linux, Mac OS X
Language Python, R
Article (Boegel et al., 2013) HLA typing from RNA-Seq sequence reads. Genome medicine.
PubMed http://www.ncbi.nlm.nih.gov/pubmed/23259685
URL https://bitbucket.org/sebastian_boegel/seq2hla
源代码,下载并安装
https://bitbucket.org/sebastian_boegel/seq2hla/src
http://tron-mainz.de/tron-facilities/computational-medicine/seq2hla/
第一版是这样的
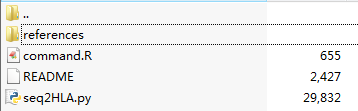
第二版是这样的
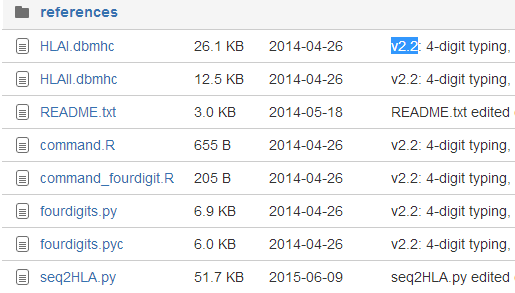
只有第二版才支持gz压缩包格式的fastq,而且不需要指定length了
其中reference文件夹下面的是发布这个软件的团体已经制备好来的HLA库文件
下载即可使用,前提是你的系统其它环境都OK
用法:
python seq2HLA.py -1 <readfile1> -2 <readfile2> -r "<runname>" [-p <int>]* [-3 <int>]**
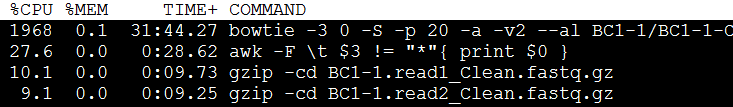
很简单,-1和-2指定我们的双端测序数据即可,可以是压缩包格式的(自动调用gzip),-r的输出目录,会输出7个文件,需要一个个解读,-p指定线程数给bowtie用的,-3是指定需要trim几个低质量碱基。
但是运行这个软件的要求非常多,需要安装好python和R,而且还有版本限制,需要安装好biopython而且还必须是双端测序,而且当前文件夹下面的reference文件夹下面必须有参考基因组的bowtie索引,而且系统必须安装好了bowtie,还需要在快捷方式里面!
我这里用的是第二版的
所以,我用的也是第二版改进的命令。非常好用,我这里用的是一个外显子测序数据,是hiseq2500测的PE100
python seq2HLA.py -1 ../../6-exon/PC3-1.read1_Clean.fastq.gz -2 ../../6-exon/PC3-1.read2_Clean.fastq.gz -r PC3
貌似输出文件太多了一点
#Output:#The results are output to stdout and to textfiles. Most important are:
#i) <prefix>-ClassI.HLAgenotype2digits => 2 digit result of Class I
#ii) <prefix>-ClassII.HLAgenotype2digits => 2 digit result of Class II
#iii) <prefix>-ClassI.HLAgenotype4digits => 4 digit result of Class I
#iv) <prefix>-ClassII.HLAgenotype4digits => 4 digit result of Class II
#v) <prefix>.ambiguity => reports typing ambuigities (more than one solution for an allele possible)
#vi) <prefix>-ClassI.expression => expression of Class I alleles
#vii) <prefix>-ClassII.expression => expression of Class II alleles
根据文献,我简单看了一下,文件的确好复杂,不过我们只需要看输出日志即可
-----------2 digit typing results-------------
#Locus Allele 1 Confidence Allele 2 Confidence
A A*68 7.287148e-05 A*24 0.03680272
B B*52 0.1717737 B*53 0.3952319
C C*12 0.03009331 hoz("C*14") 0.6783964
Calculation of locus-specific expression ...
BC1-1/BC1-1-ClassI.bowtielog
A: 7.93 RPKM
C: 9.75 RPKM
B: 8.35 RPKM
The digital haplotype is written into BC1-1/BC1-1-ClassI.digitalhaplotype3
-----------4 digit typing results-------------
#Locus Allele 1 Confidence Allele 2 Confidence
!A A*68:01 7.287148e-05 A*24:02 0.03680272
!B B*52:01 0.1717737 B*53:01' 0.6542288
!C C*12:02 0.03371717 C*12:02 0.6783964
上面的HLA的class I的数据结果
接下来是class II的数据结果,是不是很简单呀!
-----------2 digit typing results-------------
#Locus Allele 1 Confidence Allele 2 Confidence
DQA DQA1*01 0.1511134 DQA1*02 0
DQB DQB1*02 0.02321615 DQB1*05 0.42202
DRB DRB1*15 2.595144e-05 DRB1*07 0.321219
Calculation of locus-specific expression ...
BC1-1/BC1-1-ClassII.bowtielog
DQB1: 4.47 RPKM
DRB1: 5.59 RPKM
DQA1: 0.44 RPKM
-----------4 digit typing results-------------
#Locus Allele 1 Confidence Allele 2 Confidence
!DQA DQA1*01:02' 0.1511134 DQA1*02:01 0.0
!DQB DQB1*02:01' 0.02321615 DQB1*05:01 0.42202
!DRB DRB1*15:02' 2.595144e-05 DRB1*07:01 0.321219
