如果你是最近关注我们,你将又知道一个学习生信的好地方;
如果你是一直关注我们,你肯定对这个地方不陌生;
那就是我们的生信技能树论坛。
本周我们将为大家带来论坛-生信基础版块的介绍。
作者:梅零落
如果你是最近关注我们,你将又知道一个学习生信的好地方;
如果你是一直关注我们,你肯定对这个地方不陌生;
那就是我们的生信技能树论坛。
本周我们将为大家带来论坛-生信基础版块的介绍。
作者:梅零落
熟悉GSEA软件的都知道,它只需要GCT,CLS和GMT文件,其中GMT文件,GSEA的作者已经给出了一大堆!就是记录broad的Molecular Signatures Database (MSigDB) 已经收到了18026个geneset,但是我奇怪的是里面竟然没有包括cancer testis的gene set,MSigDB的确是多,但未必全,其实里面还有很多重复。而且有不少几乎没有意义的gene set。那我想做自己的gene set来用gsea软件做分析,就需要自己制造gmt格式的数据。因为即使下载了MSigDB的gene set,本质上就是gmt格式的数据而已:http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats#GMT:_Gene_Matrix_Transposed_file_format_.28.2A.gmt.29 Continue reading
这本是我为论坛的基础板块写的一个基础知识点,但是浏览量实在有限,不忍它蒙尘,特在博客重新发布一次!原帖见:http://www.biotrainee.com/thread-511-1-1.html
gene symbol 是非常官方的,由HUGO 组织负责维护,有专门的数据库HGNC database of human gene names | HUGO
以前分析数据的时候,有一些基因的symbol很奇怪,让我百思不得其解,比如
C orf 系列基因,
HS.系列基因,
KRTAP系列基因,
LOC系列基因,
MIR系列基因,
LINC系列基因
它们往往一个系列,就有好几百个基因;
C12orf44; Chromosome 12 Open Reading Frame 44; 这个是C orf系列基因的意思
MIR系列基因应该是 miRNA相关的基因
LINC系列基因应该就是long intergenic non-protein coding RNA
LOC系列基因,是非正式的,推定的,日后可能被更合适的名字替代
我这里做好了所有的基因对应关系,去生信菜鸟团QQ群里下载吧,共47938个基因的symbol和entrez gene id还有name,还有alias的对应!
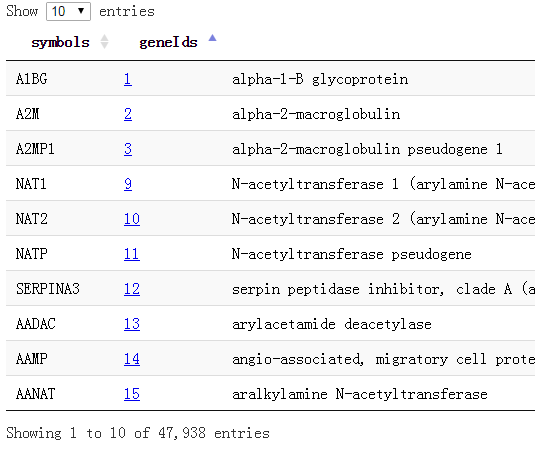
还有一些RNA基因,根本就没有symbol,比如:CTA/B/C/D系列的
Aliases for ENSG00000271971 Gene
Quality Score for this RNA gene is 1
Aliases for ENSG00000271971 Gene
CTD-2006H14.2 5
External Ids for ENSG00000271971 Gene
Ensembl: ENSG00000271971
还有,如果你看到HS.开头的基因,它是unigene的ID了,已经不再是symbol啦。
很久以前就有人问过这个问题啦,虽然目前主流还是用RPKM/FPKM来形容一个基因的表达量。但是既然大家都说TPM更好,我也来探究一下吧!
我不喜欢看公式,直接说事情,我有一个基因A,它在这个样本的转录组数据中被测序而且mapping到基因组了 5000个的reads,而这个基因A长度是10K,我们总测序文库是50M,所以这个基因A的RPKM值是 5000除以10,再除以50,为10. 就是把基因的reads数量根据基因长度和样本测序文库来normalization 。 Continue reading
看了illumina的测序仪市场份额的确很夸张,像我这样在生信数据分析领域身经百战的老鸟,都是直到今天才碰到color space的测序数据。测序平台是AB 5500xl Genetic Analyzer,就是传说中的solid格式。主要是我在学习一篇关于tp53转录因子结合能力的文章的时候碰到的 ,我查看了下载的数据虽然还是fastq格式,但很诡异,我完全不认识里面的序列。这里总结一下,下面是我的学习过程及思路,有点乱,大家随便看看!
在R里面实现这个功能其实非常简单,难的是很多packages经常会出现安装问题,更有的人压根不看芯片平台是什么,芯片对应的package是什么,就开始到处发问,自学能力实在是堪忧!
我前面有写目前所有bioconductor支持的芯片平台对应关系:通过bioconductor包来获取所有的芯片探针与gene的对应关系
但那其实是一个很笨的办法,得到所有的各式各样的探针ID与基因的对应关系,以为它绕路了,正常情况只需要在GEO里面找到芯片对应基因关系即可,没必要下载那么多package的,但是这样做的好处也是很明显的, 对很多初学者来说,如果package能解决的话,就省心很多,比如下面这个转换关系:
suppressPackageStartupMessages(library(CLL))## 这个package自带了一个数据,是我们需要用的data(sCLLex) ## 这个数据里面有24个样本,分成两组,可以直接拿来测试差异基因分析library(hgu95av2.db) ## 一定要搞清楚自己的芯片是什么数据包exprSet=exprs(sCLLex) ##得到表达数据矩阵,但是矩阵的行名,是探针ID,无法理解,需要转换##首先你取出所有的探针ID,#这里可以用三种方法来得到symbol,或者得到entrezID也可以probeset=rownames(exprSet)Symbol=as.character(as.list(hgu95av2SYMBOL[probeset]))#annotate包提供 getSYMBOL( probeset ,"hgu95av2" )#还可以用lookUp函数 lookUp( probeset , "hgu95av2", "SYMBOL")#这些只是技巧而已啦a=cbind.data.frame(Symbol,exprSet)## 下面这个函数是对每个基因挑选最大表达量探针rmDupID <-function(a=matrix(c(1,1:5,2,2:6,2,3:7),ncol=6)){exprSet=a[,-1]rowMeans=apply(exprSet,1,function(x) mean(as.numeric(x),na.rm=T))a=a[order(rowMeans,decreasing=T),]exprSet=a[!duplicated(a[,1]),]#exprSet=apply(exprSet,2,as.numeric)exprSet=exprSet[!is.na(exprSet[,1]),]rownames(exprSet)=exprSet[,1]exprSet=exprSet[,-1]return(exprSet)}exprSet=rmDupID(a)
我是受到了SOAPfuse的启发才想到整理各种基因组版本的对应关系,完整版!!!
以后再也不用担心各种基因组版本混乱了,我还特意把所有的下载链接都找到了,可以下载任意版本基因组的基因fasta文件,gtf注释文件等等!!!
GRCh36 (hg18): ENSEMBL release_52.GRCh37 (hg19): ENSEMBL release_59/61/64/68/69/75.GRCh38 (hg38): ENSEMBL release_76/77/78/80/81/82.
Feb 13 2014 00:00 Directory April_14_2003 Apr 06 2006 00:00 Directory BUILD.33 Apr 06 2006 00:00 Directory BUILD.34.1 Apr 06 2006 00:00 Directory BUILD.34.2 Apr 06 2006 00:00 Directory BUILD.34.3 Apr 06 2006 00:00 Directory BUILD.35.1 Aug 03 2009 00:00 Directory BUILD.36.1 Aug 03 2009 00:00 Directory BUILD.36.2 Sep 04 2012 00:00 Directory BUILD.36.3 Jun 30 2011 00:00 Directory BUILD.37.1 Sep 07 2011 00:00 Directory BUILD.37.2 Dec 12 2012 00:00 Directory BUILD.37.3
1. Navigate to http://genome.ucsc.edu/cgi-bin/hgTables2. Select the following options:
clade: Mammal
genome: Human
assembly: Feb. 2009 (GRCh37/hg19)
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
region: Select "genome" for the entire genome.
output format: GTF - gene transfer format
output file: enter a file name to save your results to a file, or leave blank to display results in the browser3. Click 'get output'.
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;## 这里也可以用NCBI的:ftp://ftp.ncbi.nih.gov/genomes/M_musculus/ARCHIVE/MGSCv3_Release3/Assembled_Chromosomes/chr前缀
done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta
现有的基因芯片种类不要太多了!
gpl organism bioc_package1 GPL32 Mus musculus mgu74a2 GPL33 Mus musculus mgu74b3 GPL34 Mus musculus mgu74c6 GPL74 Homo sapiens hcg1107 GPL75 Mus musculus mu11ksuba8 GPL76 Mus musculus mu11ksubb9 GPL77 Mus musculus mu19ksuba10 GPL78 Mus musculus mu19ksubb11 GPL79 Mus musculus mu19ksubc12 GPL80 Homo sapiens hu680013 GPL81 Mus musculus mgu74av214 GPL82 Mus musculus mgu74bv215 GPL83 Mus musculus mgu74cv216 GPL85 Rattus norvegicus rgu34a17 GPL86 Rattus norvegicus rgu34b18 GPL87 Rattus norvegicus rgu34c19 GPL88 Rattus norvegicus rnu3420 GPL89 Rattus norvegicus rtu3422 GPL91 Homo sapiens hgu95av223 GPL92 Homo sapiens hgu95b24 GPL93 Homo sapiens hgu95c25 GPL94 Homo sapiens hgu95d26 GPL95 Homo sapiens hgu95e27 GPL96 Homo sapiens hgu133a28 GPL97 Homo sapiens hgu133b29 GPL98 Homo sapiens hu35ksuba30 GPL99 Homo sapiens hu35ksubb31 GPL100 Homo sapiens hu35ksubc32 GPL101 Homo sapiens hu35ksubd36 GPL201 Homo sapiens hgfocus37 GPL339 Mus musculus moe430a38 GPL340 Mus musculus mouse430239 GPL341 Rattus norvegicus rae230a40 GPL342 Rattus norvegicus rae230b41 GPL570 Homo sapiens hgu133plus242 GPL571 Homo sapiens hgu133a243 GPL886 Homo sapiens hgug4111a44 GPL887 Homo sapiens hgug4110b45 GPL1261 Mus musculus mouse430a249 GPL1352 Homo sapiens u133x3p50 GPL1355 Rattus norvegicus rat230251 GPL1708 Homo sapiens hgug4112a54 GPL2891 Homo sapiens h20kcod55 GPL2898 Rattus norvegicus adme16cod60 GPL3921 Homo sapiens hthgu133a63 GPL4191 Homo sapiens h10kcod64 GPL5689 Homo sapiens hgug4100a65 GPL6097 Homo sapiens illuminaHumanv166 GPL6102 Homo sapiens illuminaHumanv267 GPL6244 Homo sapiens hugene10sttranscriptcluster68 GPL6947 Homo sapiens illuminaHumanv369 GPL8300 Homo sapiens hgu95av270 GPL8490 Homo sapiens IlluminaHumanMethylation27k71 GPL10558 Homo sapiens illuminaHumanv472 GPL11532 Homo sapiens hugene11sttranscriptcluster73 GPL13497 Homo sapiens HsAgilentDesign02665274 GPL13534 Homo sapiens IlluminaHumanMethylation450k75 GPL13667 Homo sapiens hgu21976 GPL15380 Homo sapiens GGHumanMethCancerPanelv177 GPL15396 Homo sapiens hthgu133b78 GPL17897 Homo sapiens hthgu133a
gpl_info=read.csv("GPL_info.csv",stringsAsFactors = F)### first download all of the annotation packages from bioconductorfor (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform) ##主要是因为我处理包的字符串前面有空格#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) == FALSE) {BiocInstaller::biocLite(platformDB)#source("http://bioconductor.org/biocLite.R");#biocLite(platformDB )}}
下载完了所有的包, 就可以进行批量导出芯片探针与gene的对应关系!
for (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform)#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) != FALSE) {library(platformDB,character.only = T)#tmp=paste('head(mappedkeys(',platform,'ENTREZID))',sep='')#eval(parse(text = tmp))###重点在这里,把字符串当做命令运行all_probe=eval(parse(text = paste('mappedkeys(',platform,'ENTREZID)',sep='')))EGID <- as.numeric(lookUp(all_probe, platformDB, "ENTREZID"))##自己把内容写出来即可}}
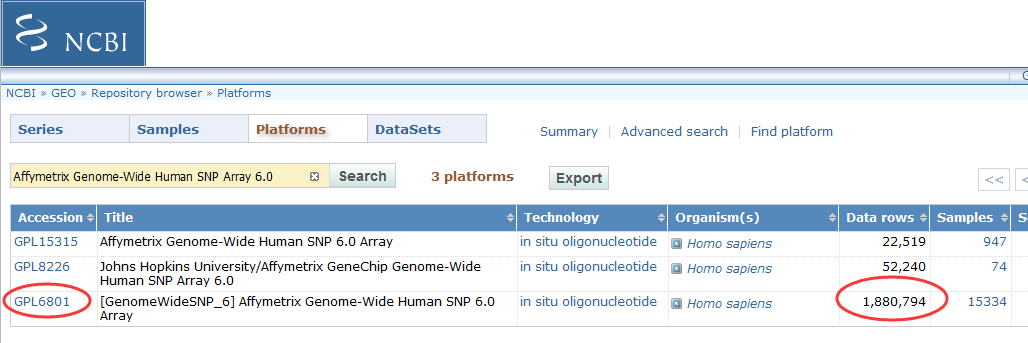
产品特点:
1.涵盖超过1,800,000个遗传变异标志物:包括超过906,600个SNP和超过946,000个用于检测拷贝数变化(CNV,Copy Number Variation)的探针;
2.SNP和CNV两种探针高密度且均匀地分布在整个基因组,不仅可以用于SNP基因精确分型,还可用于拷贝数变异CNV的研究;
3.744,000个探针平均分配到整个基因组上,用来发现未知的拷贝数变异区域;
4.可用于Copy-neutral LOH/UPD检测,亲子鉴定,纯合性分析、血缘关系鉴定、遗传病或其它疾病的研究。

chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0 chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
我的数据如下,需要自己创建成一个GRanges对象
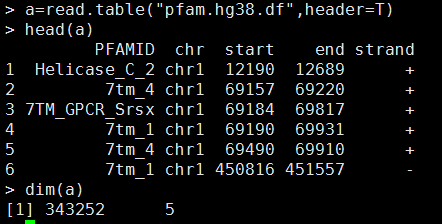
library(GenomicRanges)
pfam.hg38 <- GRanges(seqnames=Rle(a[,2]),
ranges=IRanges(a[,3], a[,4]),
strand=a[,5])
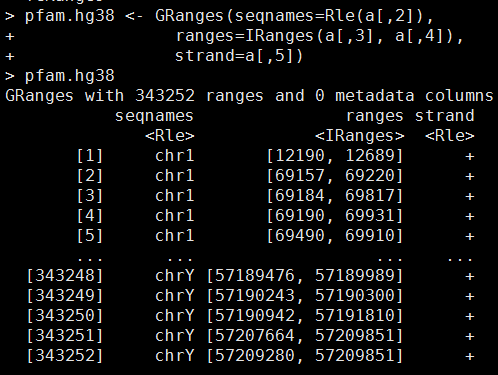
library(rtracklayer)
ch = import.chain("hg38ToHg19.over.chain")
pfam.hg19 = liftOver(pfam.hg38, ch)
前面我已经讲了如何用annovar来把vcf格式的snp进行注释,注释之后大概是这样的,每个snp位点的坐标,已经在哪个基因上面,都标的很清楚啦,。而且该突变是在哪个基因的哪个转录本的哪个外显子都一清二楚,更强大的是,还能显示是第几个碱基突变成第几个,同样氨基酸的突变情况也很清楚。
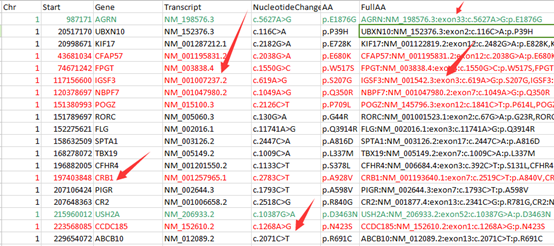
但是这样不是很方便浏览具体突变情况,所以我写了一个脚本格式化该突变情况。
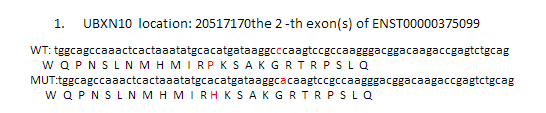
理论上是应该要做出上面这个样子,突变氨基酸前后各12个氨基酸都显示出来,突变的那个还要标红色突出显示!但是颜色控制很麻烦,我就没有做。效果如下
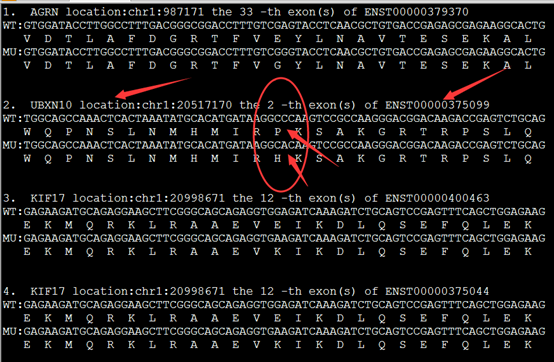
实现这样的格式化输出有三个重点,首先是NM开头的refseq的ID号要转换为ensembl数据库的转录本ID号,还有找到该转录本的CDS序列,这个都需要在biomart里面转换,或者自己写脚本,然后就用脚本爬取即可!
代码如下
[perl]
open FH1,"NM2ensembl.txt";
while(<FH1>){
chomp;
@F=split;
$hash_nm_enst{$F[4]}=$F[1] if $F[4];
}
open FH2,"ENST.CDS.fa";
while($line=<FH2>){
chomp $line;
if ($line=~/>/) {$key = (split /\|/,$line)[1];}
else {$hash_nucl{$key}.=$line;}
}
open FH3,"ENST.protein";
while($line=<FH3>){
chomp $line;
if ($line=~/>/) {$key = (split /\|/,$line)[1];}
else {$hash_prot{$key}.=$line;}
}
open FH4,"raw.mutiple.txt";
$i=1;
while(<FH4>){
chomp;
@F=split;
@tmp=split/:/,$F[1];
/:exon(\d+):/;$exon=$1;
/(NM_\d+)/; $nm=$1;
$enst=$hash_nm_enst{$nm};
print "$i. $tmp[0] $F[0] the $exon -th exon(s) of $enst \n";
$i++;
$tmp[3]=~/(\d+)/;$num_nucl=$1;
$tmp[3]=~/>([ATCG])/;$mutation_nucl=$1;
$tmp[4]=~/(\d+)/;$num_prot=$1;
$sequence=$hash_nucl{$enst};
$num_up=3*$num_prot-39;
$out_nucl=substr($sequence,$num_up,75);
print "WT:$out_nucl\n ";
for(my $j=0; $j < (length($out_nucl) - 2) ; $j += 3)
{print ' ';print $codon{substr($out_nucl,$j,3)} ;print ' ';}
print "\n";
$mutation_pos=$num_nucl-$num_up-1;
substr($out_nucl,$mutation_pos,1,$mutation_nucl) if ((length $out_nucl) == 75 );
print "MU:$out_nucl\n ";
for(my $j=0; $j < (length($out_nucl) - 2) ; $j += 3)
{print ' ';print $codon{substr($out_nucl,$j,3)} ;print ' ';}
print "\n";
print "\n";
print "\n";
}
[/perl]
http://statgenpro.psychiatry.hku.hk/limx/kggseq/download/resources/
这个网站收集了大部分资料,我们就用它的,如果它倒闭了,大家再想办法去搜索吧。
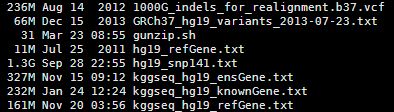
其实这些文件都是基于NCBI以及UCSC和ensembl数据库的文件用一些脚本转换而来的,都是非常简单的脚本。
首先我们看看humandb/hg19_refGene.txt 这个文件,总共2.5万多个基因的共5万多个转录本。
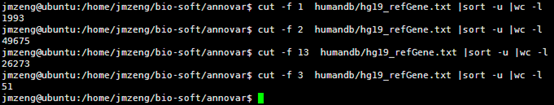
19 可能是entrez ID,但是又不像。
NM_001291929 参考基因名
chr11 染色体
-
89057521
89223909
89059923
89223852
17 89057521,89069012,89070614,89073230,89075241,89088129,89106599,89133184,89133382,89135493,89155069,89165951,89173855,89177302,89182607,89184952,89223774, 89060044,89069113,89070683,89073339,89075361,89088211,89106660,89133247,89133547,89135710,89155150,89166024,89173883,89177400,89182692,89185063,89223909,
0
NOX4 基因的英文简称,通俗名
cmpl
cmpl
2,0,0,2,2,1,0,0,0,2,2,1,0,1,0,0,0,
然后我们看看hg19_snp141.txt这个文件
1 10229 A - .
1 10229 AACCCCTAACCCTAACCCTAAACCCTA - .
1 10231 C A .
1 10231 C - .
1 10234 C T .
1 10248 A T .
1 10250 A C .
1 10250 AC - .
1 10255 A - .
1 10257 A C .
1 10259 C A .
1 10291 C T .
1 10327 T C .
1 10329 ACCCCTAACCCTAACCCTAACCCT - .
1 10330 C - .
1 10390 C - .
1 10440 C A .
1 10440 C - .
1 10469 C G .
1 10492 C T .
1 10493 C A .
1 10519 G C .
1 10583 G A 0.144169
1 10603 G A .
1 10611 C G 0.0188246
1 10617 CGCCGTTGCAAAGGCGCGCCG -
里面记录了以hg19为参考的所有的snp位点。
585
ENST00000518655 基因的ensembl ID号
chr1 + 11873 14409 14409 14409
4 基因有四个外显子
11873,12594,13402,13660, 12227,12721,13655,14409, 在基因的四个外显子的坐标
0
DDX11L1 基因的通俗英文名
none none -1,-1,-1,-1,
CTTGCCGTCAGCCTTTTCTTT·····gene的核苷酸序列
一、下载及安装软件
这个软件需要edu邮箱注册才能下载,可能是仅对科研高校开放吧。所以软件地址我就不列了。
它其实是几个perl程序,比较重要的是这个人类的数据库,snp注释必须的。
参考:http://annovar.readthedocs.org/en/latest/misc/accessory/
二,准备数据
既然是注释,那当然要有数据库啦!数据库倒是有下载地址
http://www.openbioinformatics.org/annovar/download/hg19_ALL.sites.2010_11.txt.gz
也可以用命令来下载
Perl ./annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
然后我们是对snp-calling流程跑出来的VCF文件进行注释,所以必须要有自己的VCF文件,VCF格式详解见本博客另一篇文章,或者搜索也行
http://vcftools.sourceforge.net/man_latest.html
三、运行的命令
首先把vcf格式文件,转换成空格分隔格式文件,自己写脚本也很好弄
perl convert2annovar.pl -format vcf
/home/jmzeng/raw-reads/whole-exon/snp-calling/tmp1.vcf >annovar.input
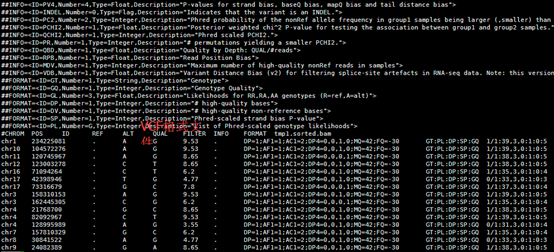
变成了空格分隔的文件
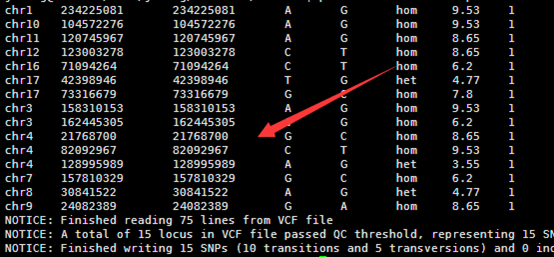
然后把转换好的数据进行注释即可
./annotate_variation.pl -out ex1 -build hg19 example/ex1.avinput humandb/
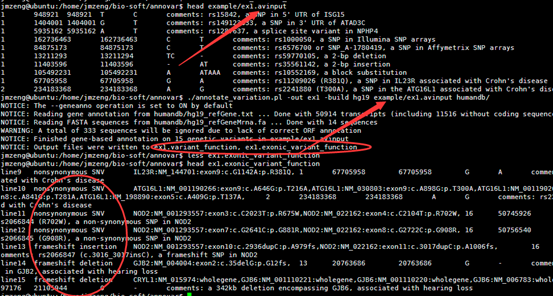
四,输出文件解读
比对可以选择BWA或者bowtie,测序数据可以是单端也可以是双端,我这里简单讲一个,但是脚本都列出来了。而且我选择的是bowtie比对,然后单端数据。
首先进入hg19的目录,对它进行两个索引
samtools faidx hg19.fa
Bowtie2-build hg19.fa hg19
我这里随便从26G的测序数据里面选取了前1000行做了一个tmp.fa文件,进入tmp.fa这个文件的目录进行操作
Bowtie的使用方法详解见http://www.bio-info-trainee.com/?p=398
bowtie2 -x ../../../ref-database/hg19 -U tmp1.fa -S tmp1.sam
samtools view -bS tmp1.sam > tmp1.bam
samtools sort tmp1.bam tmp1.sorted
samtools index tmp1.sorted.bam
samtools mpileup -d 1000 -gSDf ../../../ref-database/hg19.fa tmp1.sorted.bam |bcftools view -cvNg - >tmp1.vcf
然后就能看到我们产生的vcf变异格式文件啦!
当然,我们可能还需要对VCF文件进行再注释!
要看懂以上流程及命令,需要掌握BWA,bowtie,samtools,bcftools,
数据格式fasta,fastq,sam,vcf,pileup
如果是bwa把参考基因组索引化,然后aln得到后缀树,然后sampe对双端数据进行比对
首先bwa index 然后选择算法,进行索引。
然后aln脚本批量处理
==> bwa_aln.sh <==
while read id
do
echo $id
bwa aln hg19.fa $id >$id.sai
done <$1
然后sampe脚本批量处理
==> bwa_sampe.sh <==
while read id
do
echo $id
bwa sampe hg19.fa $id*sai $id*single >$id.sam
done <$1
然后是samtools的脚本
==> samtools.sh <==
while read id
do
echo $id
samtools view -bS $id.sam > $id.bam
samtools sort $id.bam $id.sorted
samtools index $id.sorted.bam
done <$1
然后是bcftools的脚本
==> bcftools.sh <==
while read id
do
echo $id
samtools mpileup -d 1000 -gSDf ref.fa $id*sorted.bam |bcftools view -cvNg - >$id.vcf
done <$1
==> mpileup.sh <==
while read id
do
echo $id
samtools mpileup -d 100000 -f hg19.fa $id*sorted.bam >$id.mpileup
done <$1
首先进入bowtie的主页,千万要谷歌!!!
http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
主页里面有下载链接,也有索引文件,当然索引文件只有人类等模式生物
下载之后是个压缩包,解压即可使用
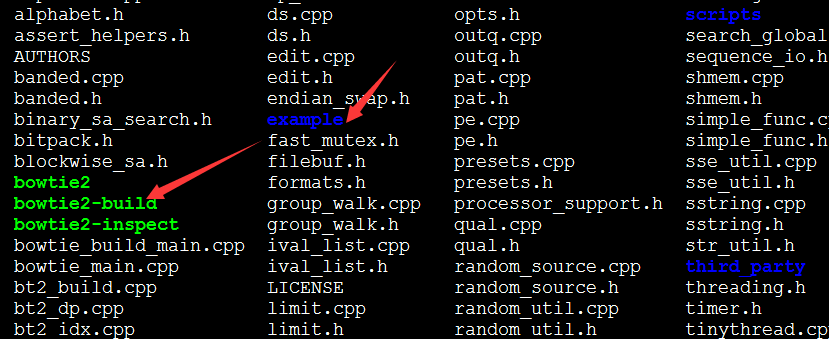
可以看到绿色的就是命令,可以添加到环境变量使用,也可以直接用全路径使用它!!!
然后example文件夹里面有所有的测试文件。
二、准备数据
我们就用软件自带的测试数据
三、运行命令
分为两步,首先索引,然后比对!!!
然后你的目录就产生了六个索引文件,我给索引取名是tmp,你们可以随便取名字
重点参数的-x 和 –S ,单端是-U 双端是-1 -2
Bowtie –x tmp –U reads.fa –S hahahhha.sam
Bowtie –x tmp -1 reads1.fa -2 reads2.fa –S hahahha.sam
四:输出文件解读
就是输出了sam文件咯,这个就看我的sam文件格式讲解哈
YGC是我非常喜欢的一个博客,但是之前都是英文,所以我没怎么看,余老师还在R领域很有建树,个人发表了5个R在生信方面的包,我后面有空会一个个试用学习。
本文转自http://ygc.name/2014/08/27/insertion-size/
里面详细解释了NGS测序的几个基本概念,也是我之前一直弄混淆的概念,包括插入片段、单端测序,双端测序,配对测序,contig,scaffold等等
在进行测序的时候,需要将DNA打断,构建library,这些fragment需要接上adaptor,好进行扩增,illumina的测序,可以有single end和paired end两种,分别从一端和两端进行测序。
fragment ======================================== fragment + adaptors ~~~========================================~~~ SE read ---------> PE reads R1---------> <---------R2 unknown gap ....................
insertion并不是指R1和R2之间的unknown gap,早在NGS之前,当我们在使用ecoli构建载体的时候,这个概念就已经形成,它是adaptors之间的序列。而unknown gap则称之为inner mate:
PE reads R1---------> <---------R2 fragment ~~~========================================~~~ insert ======================================== inner mate ....................
显然我们不希望看到大量的unknown gap,所以要制造短的fragment,而且技术不断发展,测序长度也越来越长,于是可以测通fragment:
fragment ~~~========================================~~~ insert ======================================== R1 -------------------------> R2 <----------------------- overlap :::::::::: stitched SE read --------------------------------------->
这样R1和R2就有overlap,合并一致序列,就可以得到完整的fragment,使用短的fragment,也就是insertion size比较小的library,测序的结果coverage比较大,因为我们可以测通fragment.
虽然adaptor不会被测序,但如果fragment太短,被读通了,则另一端的adaptor就会被测到。
tiny fragment ~~~~========================~~~~ insert ======================== R1 --------------------------> R2 <-------------------------- read-through !!! !!!
如果MiSeq设置正确的话,读通的adaptor是会被切除了,这样就会获得长度不一致的short reads,也可以使用N来替换adaptor序列,这样长度一样,但会在5' end看到很多N。如果没设置好,reads里含有adaptor序列,那么必须要通过软件去除,否则后续的分析都会有问题。
所以insertion size小有个好处,测序的genome coverage高,但是在进行de novo assembly的时候,有一个问题,如果基因组含有比read length还要长的重复元件时,就无法拼接,所以得到的是很多的contigs,它们之间的gap要长于insertion size且无法确定。这个问题是相当普遍的,即使是相对简单的ecoli基因组,也有一定数量的重复元件。
这个问题需要使用大的insertion size进行paired end测序来解决。
fragment + adaptors ~~~========================================~~~ PE reads R1---------> <---------R2 unknown gap ....................
在这种insertion size比较大的情况下,我们可以估计R1和R2之间的距离,只要有一个片段能够被mapped到unique position的话,那么另一个片段的大致位置就可以确定。所以为了达到好的拼接效果,长fragment的library也是必须的,它有可能给出 contigs间的相对位置。
所以理想的情况是使用multiple insert libraries,short-insert library可以保证获得足够的coverage,它可以告诉你contigs之间的序列,但信息是local的,它没办法告诉你怎么拼;而long- insert libary则可以告诉你一些相对global的信息。

在上面这个测试的数据中,加了long-insert libary虽然在coverage上没多少变化,但N50和最大的contig都显著提高,4.5Mb已经覆盖了~98%的ecoli基因组。
- See more at: http://ygc.name/2014/08/27/insertion-size/#sthash.fKmWKoaf.dpuf