目录
一:文献解读
二:NCBI搜索
三:构建脚本下载所有reads压缩包
四:用sra-toolkit解压 Continue reading
| Human Pseudogene Annotation |
GENCODE Annotation- Data: The current human pseudogene annotation is in GENCODE 21. . - Description: The GENCODE annotation of pseudogenes contains models that have been created by the Human and Vertebrate Analysis and Annotation (HAVANA) team, an expert manual annotation team at the Wellcome Trust Sanger Institute. This is informed by, and checked against, computational pseudogene predictions by thePseudoPipe and RetroFinder pipelines. PseudoPipe Output- Data: The current PseudoPipe results are on Ensembl genome release 79. . - Description: Genome-wide human pseudogene annotation predicted by PseudoPipe. PseudoPipe is a homology-based computational pipeline that searches a mammalian genome and identifies pseudogene sequences. - Reference: Other Human Pseudogene Sets- Data: . - Description: Archived pseudogene annotation on previous human genome releases from PseudoPipe. Genome-wide annotation or specific subset. |
本来搞差异分析的工具和包就一大堆了,而且limma那个包已经非常完善了,我是不准备再讲这个的,正好有个同学问了一下这个包,我就随手测试了一下,顺便看看它跟limma有什么差异没有!手痒了就记录了测试流程!
学习一个包其实非常简单,就是找到包的官网看看说明书即可!说明书链接
第一次听说这个软件,是一个香港朋友推荐的:http://davetang.org/muse/2016/01/13/getting-started-with-gemini/ 他写的很棒,但是我当初以为是一个类似于SQLite的数据库浏览模式,所以没在意。实际上,我现在仍然觉得这个软件没什么用!
软件官网有详细的介绍:https://gemini.readthedocs.io/en/latest/
而且提供丰富的教程:
We recommend that you follow these tutorials in order, as they introduce concepts that build upon one another.
软件本身并不提供注释,虽然它的功能的确包括注释,号称可以利用(ENCODE tracks, UCSC tracks, OMIM, dbSNP, KEGG, and HPRD.)对你的突变位点注释,比如你输入1 861389 . C T ,它告诉你这个突变发生在哪个基因,对蛋白改变如何?是否会产生某些疾病?
虽然它本身没有注释功能,但是它会调用snpEFF或者VEP进行注释,你需要自己先学习它们。
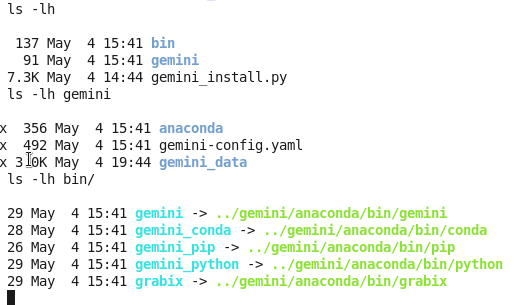
GEMINI是用python写的,有一个小脚本可以自动完成安装过程:
7.3K May 4 14:44 gemini_install.py
下载这个脚本,然后安装即可
wget https://github.com/arq5x/gemini/raw/master/gemini/scripts/gemini_install.py
python gemini_install.py $tools $data
PATH=$tools/bin:$data/anaconda/bin:$PATH
where $tools and $data are paths writable on your system.
我把$tools用的就是当前文件夹,$data也是当前文件夹下面的gemini文件夹。
这样就会在当前文件夹下面生成两个文件夹,bin是存储程序,gemini是存储数据用的,而且注意要把bin目录的全路径添加到环境变量!
我们可以直接下载软件作者提供的测试数据
首先是22号染色体的所有突变位点经过WEP注释的文件
然后是一个三口直接的突变ped格式数据
数据存放在亚马逊云,所有的教程pdf也在
http://s3.amazonaws.com/gemini-tutorials
如果是你自己的vcf文件,需要自己用VEP注释一下


产生是chr22.db就是一个数据库格式的文件,但是需要用gemini 来进行查询,个人认为,并没有多大意思!
你只要熟悉mySQL等SQL语言,完全可以自己来!
VEP是国际三大数据库之一的ENSEMBL提供的,也是非常主流和方便,但它是基于perl语言的,所以在模块方面可能会有点烦人。跟snpEFF一样,也是对遗传变异信息提供更具体的注释,而不仅仅是基于位点区域和基因。如果你熟悉外显子联盟这个数据库EXAC(ExAC.r0.3.sites.vep.vcf.gz),你可以下载它所有的突变记录数据,看看它对每个变异位点到底注释了些什么,它就是典型的用VEP来注释的。 Continue reading
这个软件比较重要,尤其是对做遗传变异相关研究的,很多人做完了snp-calling后喜欢用ANNOVAR来进行注释,但是那个注释还是相对比较简单,只能得到该突变位点在基因的哪个区域,那个基因这样的信息,如果想了解更具体一点,就需要更加功能化的软件了,snpEFF就是其中的佼佼者,而且是java平台软件,非常容易使用!而且它的手册写的非常详细:http://snpeff.sourceforge.net/SnpEff_manual.html Continue reading
string数据库是PPI领域里面最完备已经最受欢迎的数据库了。如果直接在谷歌里面搜索PPI,映入眼帘就是string的官网,它们的主页现在是html5啦,比较精美: http://string-db.org/
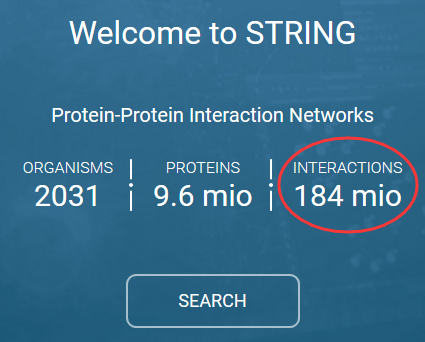
写的很霸气,近两亿的记录,不过一般大家只会关心一个物种,比如人,其实还不到一千万!
我们直接进入下载界面,找到人类的数据,人类的物种ID是9606.
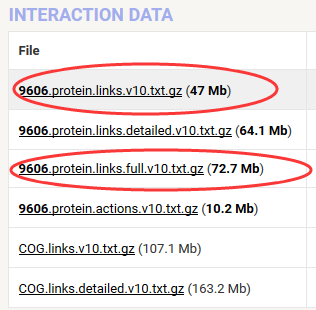
需要一定许可才能下载完整版本,我这里测试最上面那个公开版本数据!
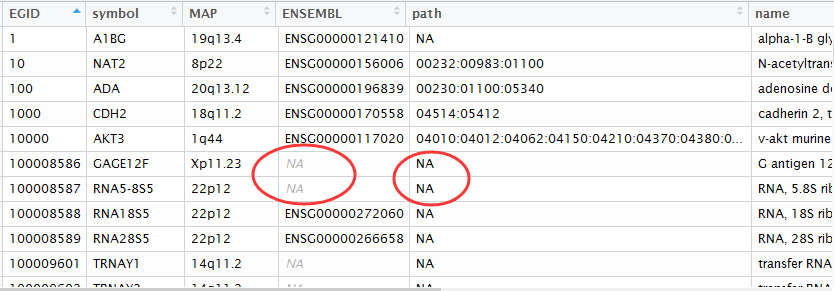
数据很简单,就是protein+protein+score,共八百多万行记录,记录着string数据库搜集的所有可能以及可信的蛋白相互作用!但是它的蛋白ID是ENSEMBL的ID,所以需要转换成基因的ID,才能被大多数人使用,因为大家的研究单位一般是基因,所以蛋白相互作用略等于基因相互作用。
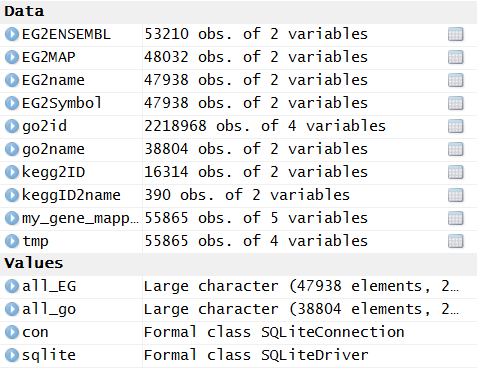
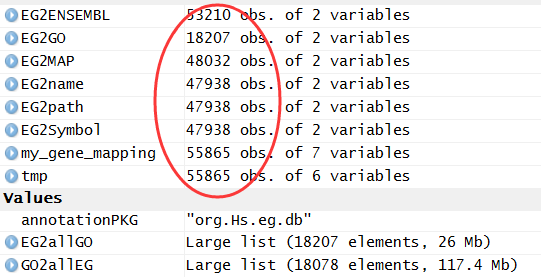
基因ID转换,我推荐用org.Hs.eg.db这个R的包,很容易就可以实现的!
> tmp=toTable(org.Hs.egENSEMBLPROT) > dim(tmp) [1] 110916 2 > head(tmp) gene_id prot_id 1 1 ENSP00000263100 2 1 ENSP00000470909 3 2 ENSP00000443302 4 2 ENSP00000323929 5 2 ENSP00000438599 6 2 ENSP00000445717 |
|
|
有约500多个蛋白ID是无法转换成对应的基因的,这个很正常,毕竟这种ID本来就不稳定,很多用着用着就失效了!
转换好之后就可以上传到数据库啦,然后可以供其它可视化或者分析程序使用!
大家分析生物信息学数据的时候必不可少的步骤就是利用各种公共资源对自己的数据进行注释。
这时候可能会用到mysql,把一些公共数据库本地化,方便使用,但是数据的下载已经存储到mysql等数据库中间会有很多值得玩味的事情。
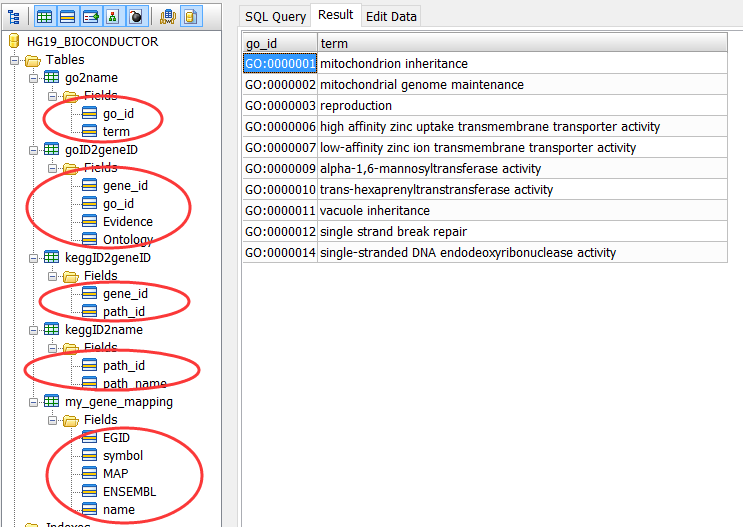
我这里给大家指出一个还算比较标准的参考,就是bioconductor官方制作的数据库设计代码。
bioconductor官方注释方面的包(主要是各种ID的转换,KEGG或者GO这样的功能注释,基因信息注释,转录本,外显子起始终止等等)
目前为止,bioconductor是3.3版本,共896个包
大部分包都是以sqlite的数据库标准发布,所以建表语句是一样的。
所有代码见:https://github.com/Bioconductor-mirror/AnnotationDbi/blob/release-3.2/inst/DBschemas
部分代码如下:
CREATE TABLE metadata (
name VARCHAR(80) PRIMARY KEY,
value VARCHAR(255)
);
CREATE TABLE go_ontology (
ontology VARCHAR(9) PRIMARY KEY, -- GO ontology (short label)
term_type VARCHAR(18) NOT NULL UNIQUE -- GO ontology (full label)
);
CREATE TABLE go_term (
_id INTEGER PRIMARY KEY,
go_id CHAR(10) NOT NULL UNIQUE, -- GO ID
term VARCHAR(255) NOT NULL, -- textual label for the GO term
ontology VARCHAR(9) NOT NULL, -- REFERENCES go_ontology
definition TEXT NULL, -- textual definition for the GO term
FOREIGN KEY (ontology) REFERENCES go_ontology (ontology)
);
CREATE TABLE sqlite_stat1(tbl,idx,stat);
CREATE TABLE go_obsolete (
go_id CHAR(10) PRIMARY KEY, -- GO ID
term VARCHAR(255) NOT NULL, -- textual label for the GO term
ontology VARCHAR(9) NOT NULL, -- REFERENCES go_ontology
definition TEXT NULL, -- textual definition for the GO term
FOREIGN KEY (ontology) REFERENCES go_ontology (ontology)
);
我是受到了SOAPfuse的启发才想到整理各种基因组版本的对应关系,完整版!!!
以后再也不用担心各种基因组版本混乱了,我还特意把所有的下载链接都找到了,可以下载任意版本基因组的基因fasta文件,gtf注释文件等等!!!
GRCh36 (hg18): ENSEMBL release_52.GRCh37 (hg19): ENSEMBL release_59/61/64/68/69/75.GRCh38 (hg38): ENSEMBL release_76/77/78/80/81/82.
Feb 13 2014 00:00 Directory April_14_2003 Apr 06 2006 00:00 Directory BUILD.33 Apr 06 2006 00:00 Directory BUILD.34.1 Apr 06 2006 00:00 Directory BUILD.34.2 Apr 06 2006 00:00 Directory BUILD.34.3 Apr 06 2006 00:00 Directory BUILD.35.1 Aug 03 2009 00:00 Directory BUILD.36.1 Aug 03 2009 00:00 Directory BUILD.36.2 Sep 04 2012 00:00 Directory BUILD.36.3 Jun 30 2011 00:00 Directory BUILD.37.1 Sep 07 2011 00:00 Directory BUILD.37.2 Dec 12 2012 00:00 Directory BUILD.37.3
1. Navigate to http://genome.ucsc.edu/cgi-bin/hgTables2. Select the following options:
clade: Mammal
genome: Human
assembly: Feb. 2009 (GRCh37/hg19)
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
region: Select "genome" for the entire genome.
output format: GTF - gene transfer format
output file: enter a file name to save your results to a file, or leave blank to display results in the browser3. Click 'get output'.
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;## 这里也可以用NCBI的:ftp://ftp.ncbi.nih.gov/genomes/M_musculus/ARCHIVE/MGSCv3_Release3/Assembled_Chromosomes/chr前缀
done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta
library(org.Hs.eg.db )
[perl]
suppressMessages(library(ggplot2))
suppressMessages(library(RMySQL))
con <- dbConnect(MySQL(), host="127.0.0.1", port=3306, user="root", password="11111111")
dbSendQuery(con, "USE test")
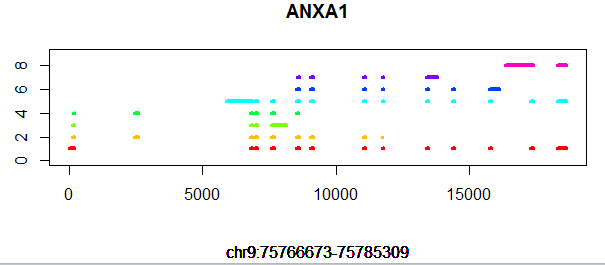
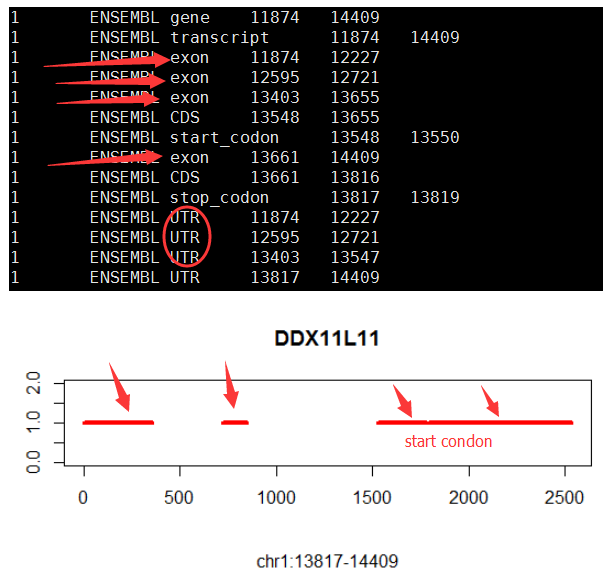
gene='SOX10'
#gene='DDX11L11'
if (T){
query=paste("select * from hg19_gtf where gene_type='protein_coding' and gene_name=",shQuote(gene),sep="")
structure=dbGetQuery(con,query)
tmp_min=min(c(structure$start,structure$end))
structure$new_start=structure$start-tmp_min
structure$new_end=structure$end-tmp_min
tmp_max=max(c(structure$new_start,structure$new_end))
num_transcripts=nrow(structure[structure$record=='transcript',])
tmp_color=rainbow(num_transcripts)
x=1:tmp_max;y=rep(num_transcripts,length(x))
#x=10000:17000;y=rep(num_transcripts,length(x))
plot(x,y,type = 'n',xlab='',ylab = '',ylim = c(0,num_transcripts+1))
title(main = gene,sub = paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
j=0;
tmp_legend=c()
for (i in 1:nrow(structure)){
tmp=structure[i,]
if(tmp$record == 'transcript'){
j=j+1
tmp_legend=c(tmp_legend,paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
}
if(tmp$record == 'exon') lines(c(tmp$new_start,tmp$new_end),c(j,j),col=tmp_color[j],lwd=4)
}
# legend('topleft',legend=tmp_legend,lty=1,lwd = 4,col = tmp_color);
}
[/perl]
现有的基因芯片种类不要太多了!
gpl organism bioc_package1 GPL32 Mus musculus mgu74a2 GPL33 Mus musculus mgu74b3 GPL34 Mus musculus mgu74c6 GPL74 Homo sapiens hcg1107 GPL75 Mus musculus mu11ksuba8 GPL76 Mus musculus mu11ksubb9 GPL77 Mus musculus mu19ksuba10 GPL78 Mus musculus mu19ksubb11 GPL79 Mus musculus mu19ksubc12 GPL80 Homo sapiens hu680013 GPL81 Mus musculus mgu74av214 GPL82 Mus musculus mgu74bv215 GPL83 Mus musculus mgu74cv216 GPL85 Rattus norvegicus rgu34a17 GPL86 Rattus norvegicus rgu34b18 GPL87 Rattus norvegicus rgu34c19 GPL88 Rattus norvegicus rnu3420 GPL89 Rattus norvegicus rtu3422 GPL91 Homo sapiens hgu95av223 GPL92 Homo sapiens hgu95b24 GPL93 Homo sapiens hgu95c25 GPL94 Homo sapiens hgu95d26 GPL95 Homo sapiens hgu95e27 GPL96 Homo sapiens hgu133a28 GPL97 Homo sapiens hgu133b29 GPL98 Homo sapiens hu35ksuba30 GPL99 Homo sapiens hu35ksubb31 GPL100 Homo sapiens hu35ksubc32 GPL101 Homo sapiens hu35ksubd36 GPL201 Homo sapiens hgfocus37 GPL339 Mus musculus moe430a38 GPL340 Mus musculus mouse430239 GPL341 Rattus norvegicus rae230a40 GPL342 Rattus norvegicus rae230b41 GPL570 Homo sapiens hgu133plus242 GPL571 Homo sapiens hgu133a243 GPL886 Homo sapiens hgug4111a44 GPL887 Homo sapiens hgug4110b45 GPL1261 Mus musculus mouse430a249 GPL1352 Homo sapiens u133x3p50 GPL1355 Rattus norvegicus rat230251 GPL1708 Homo sapiens hgug4112a54 GPL2891 Homo sapiens h20kcod55 GPL2898 Rattus norvegicus adme16cod60 GPL3921 Homo sapiens hthgu133a63 GPL4191 Homo sapiens h10kcod64 GPL5689 Homo sapiens hgug4100a65 GPL6097 Homo sapiens illuminaHumanv166 GPL6102 Homo sapiens illuminaHumanv267 GPL6244 Homo sapiens hugene10sttranscriptcluster68 GPL6947 Homo sapiens illuminaHumanv369 GPL8300 Homo sapiens hgu95av270 GPL8490 Homo sapiens IlluminaHumanMethylation27k71 GPL10558 Homo sapiens illuminaHumanv472 GPL11532 Homo sapiens hugene11sttranscriptcluster73 GPL13497 Homo sapiens HsAgilentDesign02665274 GPL13534 Homo sapiens IlluminaHumanMethylation450k75 GPL13667 Homo sapiens hgu21976 GPL15380 Homo sapiens GGHumanMethCancerPanelv177 GPL15396 Homo sapiens hthgu133b78 GPL17897 Homo sapiens hthgu133a
gpl_info=read.csv("GPL_info.csv",stringsAsFactors = F)### first download all of the annotation packages from bioconductorfor (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform) ##主要是因为我处理包的字符串前面有空格#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) == FALSE) {BiocInstaller::biocLite(platformDB)#source("http://bioconductor.org/biocLite.R");#biocLite(platformDB )}}
下载完了所有的包, 就可以进行批量导出芯片探针与gene的对应关系!
for (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform)#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) != FALSE) {library(platformDB,character.only = T)#tmp=paste('head(mappedkeys(',platform,'ENTREZID))',sep='')#eval(parse(text = tmp))###重点在这里,把字符串当做命令运行all_probe=eval(parse(text = paste('mappedkeys(',platform,'ENTREZID)',sep='')))EGID <- as.numeric(lookUp(all_probe, platformDB, "ENTREZID"))##自己把内容写出来即可}}
A general framework for estimating the relative pathogenicity of human genetic variants.
Nat Genet. 2014 Feb 2. doi: 10.1038/ng.2892.
PubMed PMID: 24487276.
Your search returned 207 results in 9 categories with the following search parameters:
| BIND | the biomolecular interaction network database | died link |
| DIP | the database of interacting proteins | http://dip.doe-mbi.ucla.edu/ |
| MINT | the molecular interaction database | http://mint.bio.uniroma2.it/mint/ |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins | http://string-db.org/ |
| HPRO | Human protein reference database | http://www.hprd.org/ |
| BioGRID | The Biological General Repository for Interaction Datasets | http://thebiogrid.org/ |
| Title | Alternatively processed and compiled RNA-Sequencing and clinical data for thousands of samples from The Cancer Genome Atlas |
| Organism | Homo sapiens |
| Experiment type | Expression profiling by high throughput sequencing |
| Summary | We reprocessed RNA-Seq data for 9264 tumor samples and 741 normal samples across 24 cancer types from The Cancer Genome Atlas with "Rsubread". Rsubread is an open source R package that has shown high concordance with other existing methods of alignment and summarization, but is simple to use and takes significantly less time to process data. Additionally, we provide clinical variables publicly available as of May 20, 2015 for the tumor samples where the TCGA ids are matched. |
前面我讲到TCGA的数据可以在5个组织机构可以获取,他们都提供了类似的接口来供用户下载数据
每个接口都有使用教程,比如http://firebrowse.org/tutorial/FireBrowse-Tutorial.pdf
非常详细!!!
有的还专门写了软件接口:https://confluence.broadinstitute.org/display/GDAC/Download
或者写了R的接口:http://www.cbioportal.org/cgds_r.jsp
接下来我们要讲的就是cbioportal网站提供的一个R接口,非常好用,只需记住4个函数即可!!! Continue reading