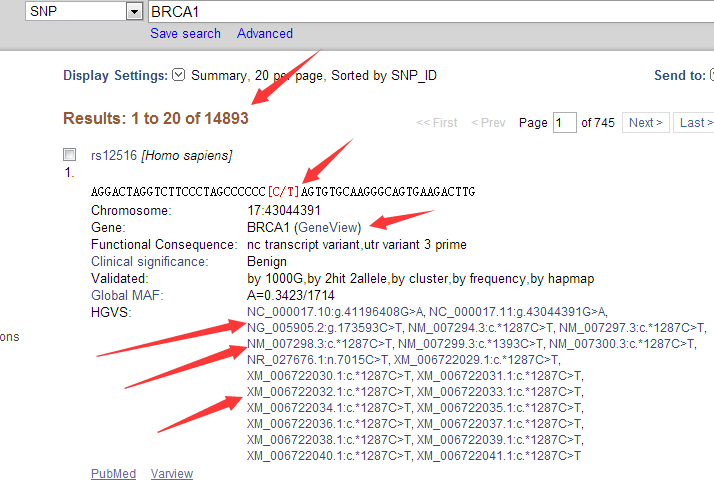
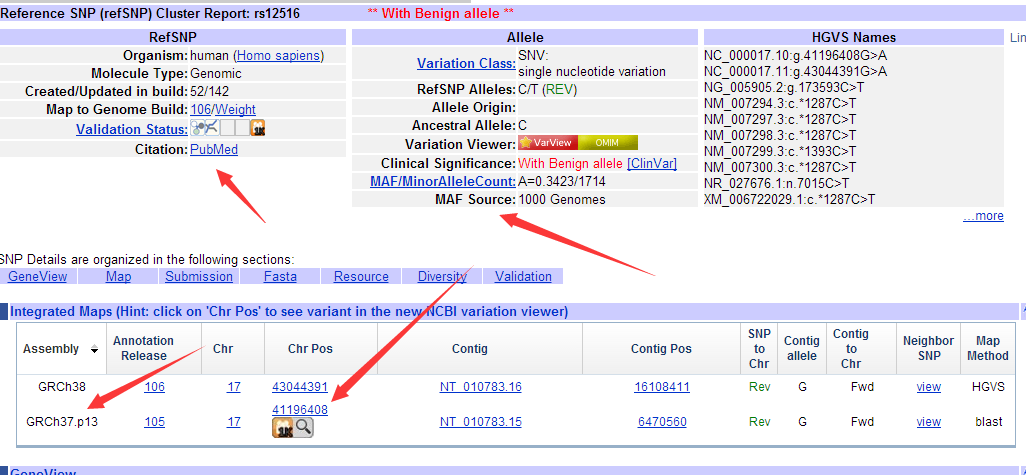
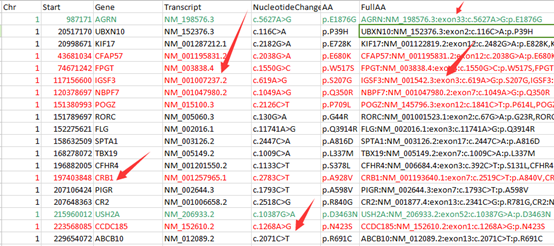
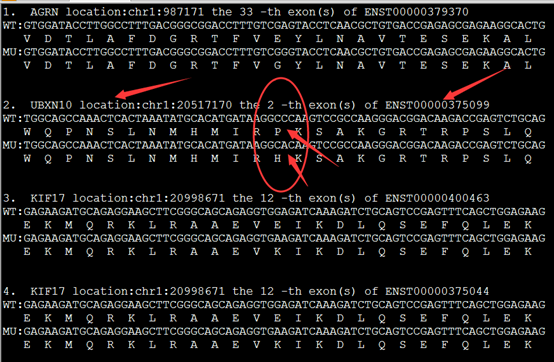
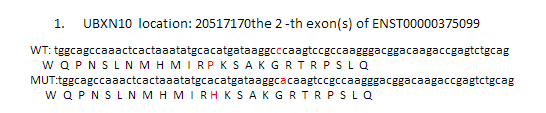并不是所有的数据都能下载,很多数据需要有权限才能下载的!!!
首先我们可以根据TCGA的文章来下载数据:
总共也就几十篇文章,都是发表在大杂志上面的。
每篇文章都会提供数据的打包下载,例如:
The molecular taxonomy of primary prostate cancer
Cell Volume 163 Issue 4: p1011-1025 Read the full article
Portal Publication Site and Associated Data Files
Comprehensive Molecular Characterization of Papillary Renal Cell Carcinoma
NEJM. Published on line on Nov 4th, 2015 Read the full article
Portal Publication Site and Associated Data Files
那个portal链接点击进去,就可以看到所有的下载链接了!
这是根据文章来分类打包好的数据!
同时也可以通过其它数据接口来下载
Tools for Exploring Data and Analyses
- Broad Institute FireBrowse portal, The Broad Institute
- cBioPortal for Cancer Genomics, Memorial Sloan-Kettering Cancer Center
- TCGA Batch Effects, MD Anderson Cancer Center
- Regulome Explorer, Institute for Systems Biology
- Next-Generation Clustered Heat Maps, MD Anderson Cancer Center
TCGA Data Portal
这几个接口都挺好用的:
非常详细!!!而且还专门写了软件接口:https://confluence.broadinstitute.org/display/GDAC/Download
或者写了R的接口:http://www.cbioportal.org/cgds_r.jsp
一般都推荐用TCGA自己的数据接口:https://tcga-data.nci.nih.gov/tcga/
里面对所有的样本都进行了统计
通过https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm可以进行定制化的数据下载!
里面有很多TCGA自定义的名词:
Data Levels and Data Types
https://tcga-data.nci.nih.gov/docs/dictionary/ 可以看到所有名词的解释:
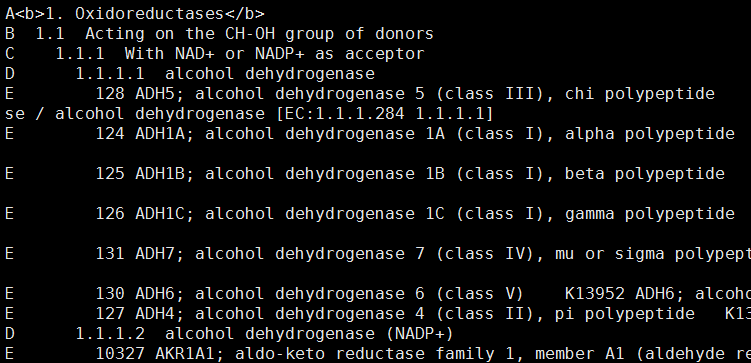
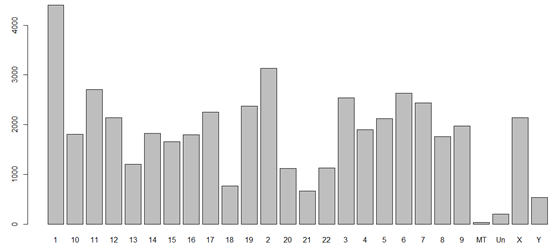
数据的种类如下:
还记得以前看到一篇TCGA自己的关于胃癌的文章,发表在nature上面,文章涉及到了TCGA的各个方面的分析,所以附件PDF竟然有133页!!!
设计到的295个sample的list在:https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/other/publications/stad_2014/STAD.Sample_Barcodes.txt
包含的其它数据有: