一,下载该软件
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
tar xzf sratoolkit.current-centos_linux64.tar.gz
解压直接使用即可,里面有一大堆的软件,针对不同的测序仪,不同的数据 Continue reading
一,下载该软件
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
tar xzf sratoolkit.current-centos_linux64.tar.gz
解压直接使用即可,里面有一大堆的软件,针对不同的测序仪,不同的数据 Continue reading
查某个基因家族在某物种的具体信息
我很伤心,不知道是不是我写的教程还是不够人性化,一个朋友在群里面问如何知道NAC基因家族在拟南芥里面的105个基因信息,我随便给他示范了一下在人类里面如何找,希望他能触类旁通,结果他不会linux,啥生信基础都没有,我只会诱导他简单学习一下,希望他至少明白什么的taxid。所以我给了他我之前写的教程,只希望他告诉我拟南芥的taxid我就帮他把那105个基因找出来。 Continue reading
找橡胶测序数据无果
所以我只好找了他们所参考的草莓(strawberry, Fragaria vesca (2n = 2x = 14),a small genome (240 Mb),)的文章,是发表是nature genetics上面的
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3326587/
可以看到它的SRA索取号。

草莓组装结果:Over 3,200 scaffolds were assembled with an N50 of 1.3 Mb .
Over 95% (209.8 Mb) of the total sequence is represented in 272 scaffolds.
草莓基因息:Gene prediction modeling identified 34,809 genes, with most being supported by transcriptome mapping.
草莓染色体信息:Paradoxically, the small basic (x = 7) genome size of the strawberry genus, ~240 Mb,
offers substantial advantages for genomic research.
草莓来源:diploid strawberry F. vesca ssp. vesca accession Hawaii 4
(National Clonal Germplasm Repository accession # PI551572).
然后我去NCBI上面下载这三个数据
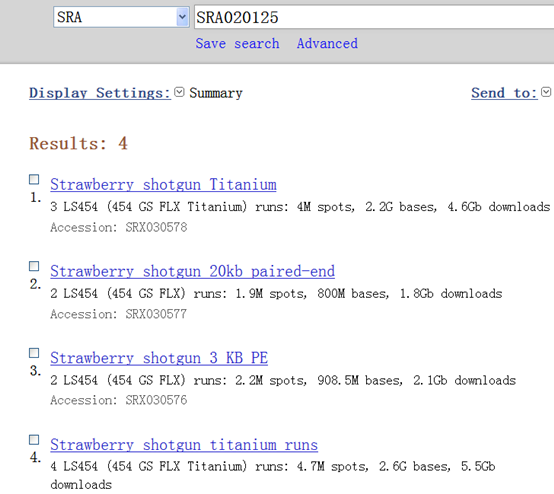
SRA020125 共有四个数据:
| http://www.ncbi.nlm.nih.gov/sra/SRX030575[accn] | Total: 4 runs, 4.7M spots, 2.6G bases, 5.5Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030576[accn] (3 KB PE) | Total: 2 runs, 2.2M spots, 908.5M bases, 2.1Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030577[accn] (20KB片段) | Total: 2 runs, 1.9M spots, 800M bases, 1.8Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030578[accn] | Total: 3 runs, 4M spots, 2.2G bases, 4.6Gb |
挂在后台自动下载
![]()
好了,有了这些数据我们就要进行基因组的一系列分析啦!!!
不过我们可以先看看他们这个研究小组的成果
首先他们建造了一个关于草莓的基因组信息网站
https://strawberry.plantandfood.co.nz/
跟我之前在水科院做鲫鱼鲤鱼的差不多
直接在里面就可以下载他们做好的所有数据,也可以可视化。
它的染色体如下,非常简单,就七条染色体
http://www.rosaceae.org/species/fragaria/fragaria_vesca/genome_v1.1
我找到了它组装好的草莓基因组地址,用批处理全部下载了

研读橡胶的基因组文章
我本科的前两年在海南儋州读书,那时候旁边就是橡胶所,很多同学也在那边做毕业论文什么的,我一直以为那里是全世界的橡胶中心,所有的先进技术都在那里产生,结果,前些天跟一个橡胶所的老师聊天才发现,居然橡胶(Hevea brasiliensis)的基因组已经发表了,可是,跟橡胶所没有半毛钱关系,更搞笑的事情是,堂堂一个基因组文章居然发表在BMC这样的杂志,真不知道是基因组的年代已经过去了还是他们做的实在是太差了,反正我看不过去了,所以研读他们的文章,并且下载数据测试一下。
文章地址如下:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3575267/

可以看到它过于数据的描述都在补充材料1里面,所以我下载了补充材料。
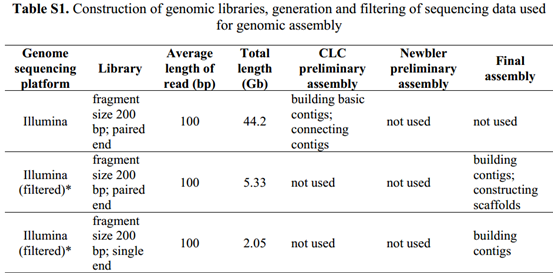
可以看到所有的测序数据的描述,45个G的i llumina的200bp的双端测序,27个G的illumina的200bp的双端测序,约10G左右的长片段(8kb,20kb)罗氏454数据,最后还有一点点solid数据,它这样的测序策略好像是模仿的2011年发布的草莓基因组数据。
但是补充材料里面没有列出下载地址,我有点困惑!
按照道理我研读文献的步骤应该没有错,有可能是因为这个文章发表的杂志水平太低,所以不要求他们把测序原始数据上传到NCBI的SRA里面。或者是他们本身觉得文章发的不够档次,不想公布数据,所以先留着自己做精细分析,等发了大文章再公布原始数据。
然后我在NCBI的SRA里面查找了关于橡胶的原始数据,果真没有
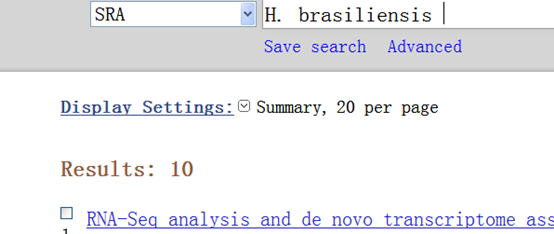
仅有的10个数据,都是别的小组做的RNA-seq的内容。
De novo transcriptome analysis of abiotic stress responsive transcripts of Hevea brasiliensis.
所以我只好找了他们所参考的草莓(strawberry, Fragaria vesca (2n = 2x = 14),a small genome (240 Mb),)的文章,是发表是nature genetics上面的
YGC是我非常喜欢的一个博客,但是之前都是英文,所以我没怎么看,余老师还在R领域很有建树,个人发表了5个R在生信方面的包,我后面有空会一个个试用学习。
本文转自http://ygc.name/2014/08/27/insertion-size/
里面详细解释了NGS测序的几个基本概念,也是我之前一直弄混淆的概念,包括插入片段、单端测序,双端测序,配对测序,contig,scaffold等等
在进行测序的时候,需要将DNA打断,构建library,这些fragment需要接上adaptor,好进行扩增,illumina的测序,可以有single end和paired end两种,分别从一端和两端进行测序。
fragment ======================================== fragment + adaptors ~~~========================================~~~ SE read ---------> PE reads R1---------> <---------R2 unknown gap ....................
insertion并不是指R1和R2之间的unknown gap,早在NGS之前,当我们在使用ecoli构建载体的时候,这个概念就已经形成,它是adaptors之间的序列。而unknown gap则称之为inner mate:
PE reads R1---------> <---------R2 fragment ~~~========================================~~~ insert ======================================== inner mate ....................
显然我们不希望看到大量的unknown gap,所以要制造短的fragment,而且技术不断发展,测序长度也越来越长,于是可以测通fragment:
fragment ~~~========================================~~~ insert ======================================== R1 -------------------------> R2 <----------------------- overlap :::::::::: stitched SE read --------------------------------------->
这样R1和R2就有overlap,合并一致序列,就可以得到完整的fragment,使用短的fragment,也就是insertion size比较小的library,测序的结果coverage比较大,因为我们可以测通fragment.
虽然adaptor不会被测序,但如果fragment太短,被读通了,则另一端的adaptor就会被测到。
tiny fragment ~~~~========================~~~~ insert ======================== R1 --------------------------> R2 <-------------------------- read-through !!! !!!
如果MiSeq设置正确的话,读通的adaptor是会被切除了,这样就会获得长度不一致的short reads,也可以使用N来替换adaptor序列,这样长度一样,但会在5' end看到很多N。如果没设置好,reads里含有adaptor序列,那么必须要通过软件去除,否则后续的分析都会有问题。
所以insertion size小有个好处,测序的genome coverage高,但是在进行de novo assembly的时候,有一个问题,如果基因组含有比read length还要长的重复元件时,就无法拼接,所以得到的是很多的contigs,它们之间的gap要长于insertion size且无法确定。这个问题是相当普遍的,即使是相对简单的ecoli基因组,也有一定数量的重复元件。
这个问题需要使用大的insertion size进行paired end测序来解决。
fragment + adaptors ~~~========================================~~~ PE reads R1---------> <---------R2 unknown gap ....................
在这种insertion size比较大的情况下,我们可以估计R1和R2之间的距离,只要有一个片段能够被mapped到unique position的话,那么另一个片段的大致位置就可以确定。所以为了达到好的拼接效果,长fragment的library也是必须的,它有可能给出 contigs间的相对位置。
所以理想的情况是使用multiple insert libraries,short-insert library可以保证获得足够的coverage,它可以告诉你contigs之间的序列,但信息是local的,它没办法告诉你怎么拼;而long- insert libary则可以告诉你一些相对global的信息。

在上面这个测试的数据中,加了long-insert libary虽然在coverage上没多少变化,但N50和最大的contig都显著提高,4.5Mb已经覆盖了~98%的ecoli基因组。
- See more at: http://ygc.name/2014/08/27/insertion-size/#sthash.fKmWKoaf.dpuf
仿写fastqc软件的一些功能(下)
文件来自于上面perl代码的输出文件,好像算法有点问题,26G的文件居然处理近一个小时才出数据!
R语言本身自带的画图工具都很丑,懒得说了,可以用ggplot2来重新画一个,不是项目要求没有报酬我就懒得画了,大家面前看看画图原理即可。
fastqc软件的使用
一:下载安装该软件
具体搜索其地址下载,fastqc是一个java软件,下载后可以直接使用,但是需要自行配置好java环境,具体配置方法,见linux下java配置。
NCBI的taxid简单介绍
物种的信息集合都在它的NCBI的taxid号里面,在NCBI里面关于它的英文介绍地址如下 http://www.ncbi.nlm.nih.gov/guide/taxonomy/ ,NCBI人为的给自然界所有的物种都给了一个编号,这个编号就是taxid,是根据计算机里面树这种数据结构来编码的,其中人类的编号是 9606,7227是果蝇,我们只需要进入这个物种的taxid里面就能看的关于它的一切NCBI存在并且收集好的信息。
NCBI的blast++软件的使用
目录
一:下载安装该软件
二:准备数据
三:运行命令
四:输出文件解读
正文
一:下载安装该软件
在NCBI的ftp站点里面可以找到blast++的下载链接
wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/ncbi-blast-2.2.30+-x64-linux.tar.gz Continue reading
搜索其他学者的RNA数据处理流程(包括原始数据、脚本、中间文件)
一:原始数据
是谷歌里面无意中搜索到的,是某个物种的RNA数据,不是很大,但是里面有所有的分析流程,非常方便,对原始reads进行了组装,和注释。
http://moana.dnsalias.org/~sgeib/Anth_RNAseq/Run2.1/RawData/
打开网址可以看到raw data的下载链接
阅读文献并下载原始数据知illumina的Chip-seq数据
目录
一:阅读文献找到总实验项目
二:在根据实验项目地址找到所有实验数据的下载地址
三:构造脚本并下载
四:用sra-toolkit工具解压
正文
一:阅读文献找到总实验项目
该chip-seq数据其实隶属于一个大的实验项目组,其下载地址如下http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52964
阅读文献并下载原始测序数据之helicos转录组数据
目录
正文
一、阅读pdf文献,并找到原始数据搜索关键词

可以看到它的下载索引是SRP003040,阅读文献可知其包含4种细胞的6种处理方式的转录组数据

